




INTRODUCTION
Continuous protein turnover occurs in the human body, wherein various proteins essential for physiological activities, such as hormones and enzymes, are synthesized while unnecessary proteins are degraded. The normal protein metabolism requires a balance between protein synthesis and degradation to maintain cellular homeostasis [1]. Certain diseases, such as cancer and neurodegenerative disorders, arise when specific proteins accumulate excessively or are improperly degraded, leading to pathological consequences [2]. Small molecule inhibitors (SMIs) have been used widely as traditional therapeutic agents to address these issues. SMIs function by inhibiting the activity of target proteins, mitigating disease progression. These inhibitors typically bind to the active site or a specific binding pocket of the protein, suppressing its function [3]. On the other hand, more than 80% of proteins are classified as “undruggable” because of the lack of well-defined binding pockets or active sites, making them challenging targets for SMIs [4]. In addition, a sustained drug concentration must be maintained to ensure therapeutic efficacy because proteins are continuously synthesized. This requirement increases the risk of adverse effects and enhances the likelihood of off-target interactions, potentially leading to unintended biological consequences [5].
Recently, therapeutic strategies have attempted to overcome these limitations using intracellular protein degradation systems to remove abnormal and misfolded proteins. The two major intracellular protein degradation pathways are the ubiquitin–proteasome system (UPS) and the autophagy–lysosome pathway [6]. The UPS functions by tagging target proteins for degradation through ubiquitination. This ubiquitin-tagged protein is then recognized by the proteasome, which then degrades it, ensuring selective protein turnover and maintaining cellular homeostasis [7]. The autophagy-lysosome pathway is a degradation mechanism that breaks down damaged organelles and protein aggregates, including large protein complexes. In this process, an autophagosome forms around the damaged proteins, encapsulating them within a double-membrane vesicle. The autophagosome then fuses with a lysosome, where hydrolytic enzymes degrade the enclosed proteins using ATP, ensuring cellular quality control and homeostasis [8]. The emerging therapeutic approach of targeted protein degradation (TPD), which leverages the intrinsic protein degradation systems of cells, enables the targeting of proteins that are traditionally considered undruggable by conventional SMIs. Unlike transient inhibition, TPD facilitates the complete removal of the target protein, offering sustained efficacy and potential for repeated therapeutic application [9, 10]. In particular, TPD can target proteins located in challenging cellular compartments, such as the extracellular matrix and cell membrane, and traditionally undruggable targets like transcription factors. This expands the scope of future research opportunities and therapeutic development [11]. Currently, various TPD technologies are being actively researched and developed, including proteolysis-targeting chimeras (PROTACs), molecular glues (MGs), autophagy-targeting chimeras (AUTOTACs), lysosome-targeting chimeras (LyTACs), and autophagy-tethering compounds (ATTECs). Among these, PROTACs and MGs have been studied extensively, with several molecules currently undergoing clinical trials [12]. This study focused on PROTACs and MGs, the two most significant approaches in the TPD field that leverage proteasome-mediated degradation. This review comprehensively explains their mechanisms, therapeutic applications, and prospects.
MECHANISM OF TARGETED PROTEIN DEGRADATION
A clear understanding of the ubiquitination process is essential for elucidating the mechanism of TPD based on the UPS. Ubiquitin is a small, evolutionarily conserved 76-amino acid protein found in all eukaryotic cells. Its final amino acid, glycine, forms a covalent bond with the lysine residue of the target proteins, marking them for degradation through the UPS [13]. Ubiquitin undergoes a sequential enzymatic cascade involving three key enzymes to be conjugated to a target protein. First, the ubiquitin-activating enzyme (E1) activates ubiquitin in an ATP-dependent manner. The ubiquitin-conjugating enzyme (E2) then transfers the activated ubiquitin from E1 to either an E3 ubiquitin ligase (E3 ligase) or directly to the substrate protein. Finally, the E3 ligase recognizes and binds to the target protein, facilitating the transfer of ubiquitin from E2 to the lysine residue of the substrate. A polyubiquitin chain is formed through this process, marking the protein for degradation via the proteasome [14]. The ubiquitinated protein is directed to the 26S proteasome through this three-step process, where it undergoes degradation, ensuring proper protein turnover and cellular homeostasis [15]. The E3 ligase is crucial in recognizing and binding to specific substrate proteins, facilitating their ubiquitination. UPS-based TPD strategies leverage these enzymes to selectively degrade target proteins because of the diversity of E3 ligases, enabling precise and efficient protein elimination [6].
First introduced in 2001, PROTACs are heterobifunctional molecules composed of separate ligands for the target protein and E3 ligase, connected by a chemical linker. This design enables the selective ubiquitination and subsequent degradation of the target protein via the UPS [16]. The target-binding ligand binds to the protein, while the E3 ligase ligand recruits an E3 ligase. Once the E3 ligase is recruited, it facilitates the ubiquitination of the target protein, marking it for proteasomal degradation via the UPS [17].
MGs are a class of small molecules that facilitate the interaction between the target protein and an E3 ligase, promoting ubiquitination and subsequent proteasomal degradation of the target protein [18, 19]. The immunomodulatory imide drug (IMiD) class, which is used as a first-line treatment for multiple myeloma, is a representative example of MGs. IMiDs interact directly with the E3 ligase cereblon (CRBN), facilitating the recruitment of neo-substrates, such as the Ikaros (IKZF) family, leading to their ubiquitination and subsequent degradation [20]. The IKZF family consists of transcription factors traditionally considered undruggable by conventional therapies. Nevertheless, IMiDs induce their polyubiquitination and subsequent proteasomal degradation, eliminating these proteins through MGs-mediated mechanisms (Fig. 1) [21].
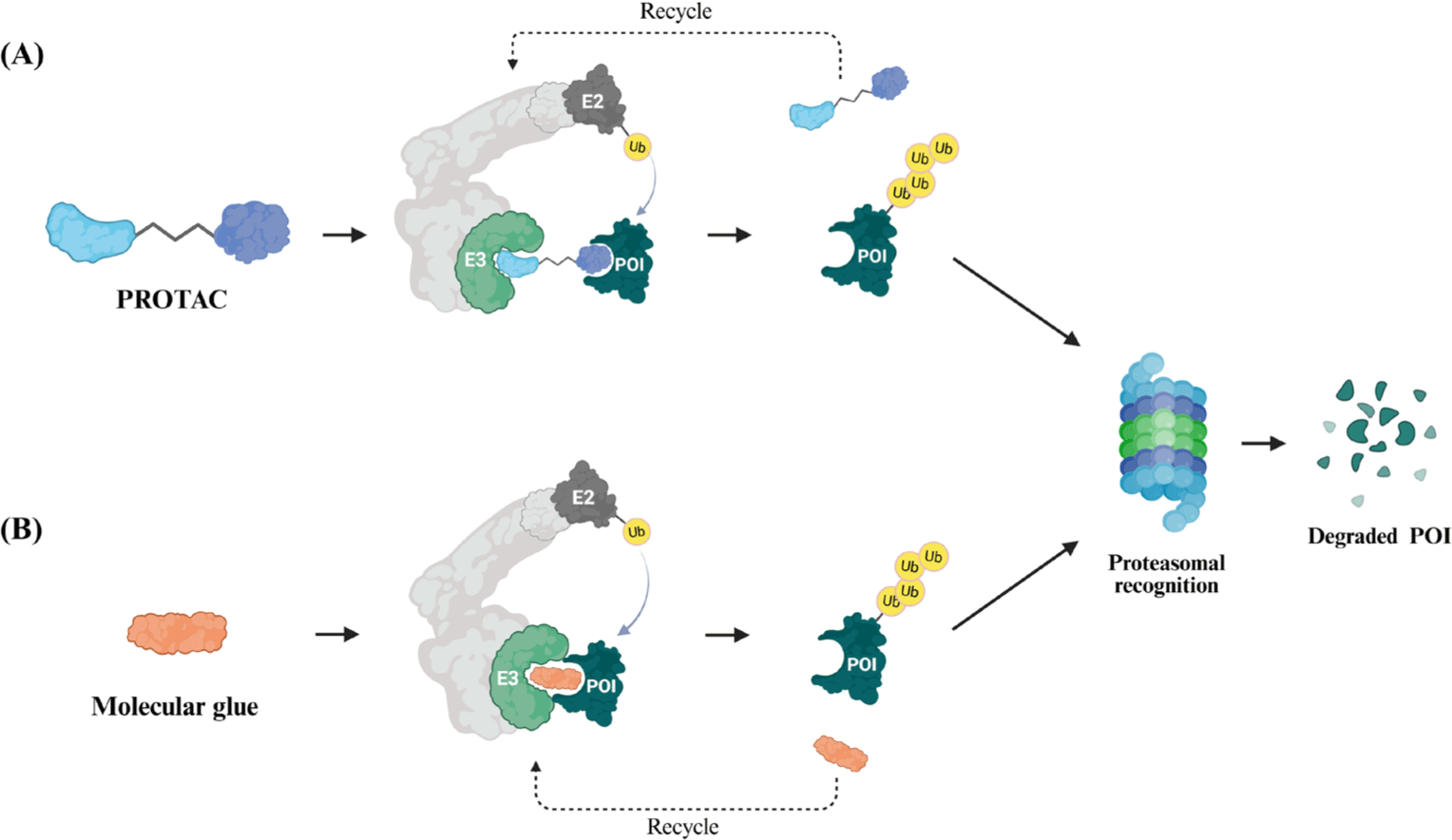
Although MGs stabilize specific protein–protein interactions (PPIs) through a single small molecule, PROTACs are heterobifunctional molecules that induce proteasomal degradation by simultaneously binding a target protein and an E3 ligase [22]. The main differences between PROTACs and MGs are summarized in Table 1. MGs generally have a low molecular weight, high cell membrane permeability, and favorable drug-like properties, making them well-suited for therapeutic applications. In contrast, PROTACs are larger molecules with complex physicochemical properties that can limit their cellular uptake and metabolic stability, posing challenges for drug delivery and bioavailability [23]. The two approaches are complementary, and MGs are often used as ligands for E3 ligases in PROTAC design, enhancing the efficiency of TPD [24–26].
In recent years, the discovery of small-molecule ligands for E3 ligases has significantly advanced the understanding and application of PROTAC technology. Most small-molecule PROTACs use Von Hippel-Lindau (VHL) and CRBN E3 ligases for TPD [27]. VHL functions as a substrate adaptor within the Cullin 2 E3 ligase complex, playing a crucial role in targeting hypoxia-inducible factor 1-alpha for rapid and efficient degradation [28, 29]. CRBN, a substrate receptor within the Cullin 4A E3 ligase complex, is the second E3 ligase commonly used in the PROTAC design [30, 31]. IMiD thalidomide and its derivatives have been shown to bind to CRBN, facilitating the degradation of various neo-substrate proteins through the UPS [32, 33]. Despite numerous E3 ligases, only 12 (approximately 1.1%) have been incorporated into PROTAC design, highlighting the need for further exploration and expansion of E3 ligase diversity in TPD strategies [34]. Several E3 ligases, including cIAP (cellular inhibitor of apoptosis protein), MDM2 (mouse double minute 2), DCAF11 (DDB1 and CUL4 associated factor 11), DCAF15, DCAF16, FEM1B (protein fem-1 homolog B), and KEAP1 (Kelch-like ECH-associated protein 1), have been reported as potential candidates for PROTAC development, expanding the range of targetable proteins in TPD strategies [35–37]. The E3 ligase library has immense untapped potential for future research. Further studies will be needed to explore new E3 ligases for TPD design, along with expanded investigations into diverse target proteins, to enhance the applicability and effectiveness of TPD-based therapies.
THERAPEUTIC APPLICATIONS OF TARGETED PROTEIN DEGRADATION
PROTAC technologies have been actively studied for various protein-related diseases, showing significant therapeutic efficacy in clinical trials. PROTACs offer a novel approach to cancer therapy by selectively degrading proteins essential for cancer cell proliferation and survival. Notable examples include ARV-110 (for prostate cancer), ARV-471 (for breast cancer), and CFT-1946 (targeting BRAF-mutant cancers) [38, 39]. In addition, PROTAC-based strategies are being explored for the removal of pathogenic protein aggregates associated with neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease [35, 40, 41]. Research is also underway to develop PROTACs targeting key proteins involved in immune response regulation, including interleukin-1 receptor-associated kinase 4, Bruton tyrosine kinase (BTK), and IKZF1/3, to modulate the inflammatory processes and provide potential therapeutic options for autoimmune and inflammatory diseases [42, 43]. The E3 ligands used in PROTACs are derived primarily from IMiDs and generally have a molecular weight of less than 300 Da. Among them, (R)-thalidomide, the E3 ligase ligand in ARV-471, has entered Phase 3 clinical trials. Thalidomide derivatives are the most commonly used CRBN ligands in PROTAC design owing to their high efficacy in recruiting E3 ligases for TPD [44]. Currently, PROTAC drugs targeting various proteins, including androgen receptor, estrogen receptor, B-cell lymphoma-extra large, IKZF1/3, and BTK, have entered clinical trial stages, highlighting their potential as novel therapeutic options (Table 2). Despite their promise, PROTAC drug development faces challenges such as complex synthesis, high costs, and regulatory uncertainties. Thus far, no PROTAC-based therapy has received approval from the Food and Drug Administration (FDA) or other regulatory agencies.
| Compound | Target protein | Indication | Reference |
|---|---|---|---|
| ARV-110 | AR | Metastatic castration-resistant prostate cancer | [65] |
| ARV-766 | AR | Metastatic castration-resistant prostate cancer | [66] |
| ARV-471 | ER | Breast cancer (ER+/HER2-) | [67] |
| DT2216 | BCL-XL | Hematologic malignancies, solid tumors | [68] |
| NX2127 | BTK, IKZF1/3 | CLL, NHL | [69] |
| NX5948 | BTK | B-cell malignant tumors and autoimmune diseases | [70] |
| CFT 1946 | Mutant BRAF V600E | Melanoma, colorectal cancer | [71] |
| CFT 8634 | Bromodomain-containing protein 9 | Synovial sarcoma | [72] |
| CFT 8919 | Mutant EGFR L858R | Non-small cell lung cancer | [73] |
| KT-474 | IRAK4 | Autoimmune diseases | [74] |
PROTACs, proteolysis-targeting chimeras; AR, androgen receptor; ER, estrogen receptor; BCL-XL, B-cell lymphoma-extra large; BTK, Bruton tyrosine kinase; IKZF, ikaros; CLL, chronic lymphocytic leukemia; NHL, non-hodgkin’s lymphoma; BRAF, B-Raf proto-oncogene, serine/threonine kinase; EGFR, epidermal growth factor receptor; IRAK4, interleukin-1 receptor-associated kinase 4.
Because MGs must induce specific PPIs, their discovery is challenging and often unpredictable, making their development more limited than other therapeutic approaches. In addition, MGs cannot be applied universally to all target proteins because they rely on interactions with specific proteins, restricting their broader applicability [23]. As a result, compared to PROTACs, MGs have attracted relatively less research and development attention. Thus far, the only FDA-approved MGs are thalidomide (Thalomid) and its analogs, lenalidomide (Revlimid) and pomalidomide (Pomalyst) [23]. Table 3 provides examples of MGs that have entered clinical trials. Bristol-Myers Squibb (Princeton, NJ, USA) is a leading company in this field, holding a pipeline of MGs through Celgene, which includes CC-92480 (IKZF1/3), CC-90009 (GSPT1), and CC-220 (IKZF1/3) [45–47]. The development of PROTAC-based and MG-based novel therapeutics is expanding steadily, and an increasing number of innovative treatments are expected to emerge.
| Complound | Target protein | Indication | Reference |
|---|---|---|---|
| CC-92480 | IKZF1/3 | MM | [75] |
| CC-90009 | GSPT1 | AML, MDS | [76] |
| CC-220 | IKZF1/3 | Relapsed/refractory MM, NHL, systemic lupus erythematosus. | [77] |
| E7820 | RBM39 | Relapsed or refractory AML, MDS or chronic myelomonocytic leukemia | [78] |
| CFT 7455 | IKZF1/3 | MM, NHL | [79] |
| NVP-DKY709 | IKZF2 | Cancer immunotherapy | [80] |
CHALLENGES AND FUTURE DIRECTIONS
TPD enables the targeting of “undruggable” proteins that were previously inaccessible to conventional therapies. Unlike traditional inhibitors that just suppress protein activity, TPD eliminates the target protein itself, allowing for complete functional removal. This approach can also reduce resistance caused by long-term selective pressure, making it a promising strategy for durable therapeutic effects [48]. In addition, unlike conventional inhibitors, TPD molecules exhibit a catalytic mode of action, enabling a single molecule to degrade multiple target proteins. This catalytic effect allows for therapeutic efficacy at lower concentrations, potentially reducing toxicity and off-target side effects and improving the overall safety profile of TPD-based therapies [6]. Nevertheless, despite their potential, PROTACs and MGs still face certain limitations that must be addressed before they can be adopted widely as therapeutics for various refractory diseases.
PROTACs face several challenges because of their relatively high molecular weight and pharmacokinetic (PK) limitations. These molecules are generally large, exhibit low solubility, and have restricted cell permeability, which can hinder their intracellular delivery. In addition, metabolic instability can lead to a short half-life in vivo, potentially limiting their therapeutic effectiveness [49]. Optimizing the design of the linker and the ligand that binds to the target protein is necessary to overcome these challenges. Enhancing linker properties can improve cell permeability, stability, and PK, while refining ligand selection can enhance the binding affinity and specificity, ultimately increasing the efficacy of PROTAC-based therapeutics [50]. Furthermore, high concentrations of PROTACs can lead to a phenomenon known as the Hook Effect, which paradoxically reduces protein degradation efficiency. This occurs when ternary complex formation between the target protein, E3 ligase, and PROTAC is impaired. Instead of forming a productive ternary complex, excess PROTAC molecules preferentially bind individually to the target protein or E3 ligase, forming an inactive binary complex and reducing degradation efficiency [51]. Careful dose optimization is essential in clinical settings to prevent this. End-point binding free energy calculation can be used to characterize the stabilization and hook effects in PROTAC systems, optimizing the computational conditions to improve the prediction accuracy for rational PROTAC design [52].
Various innovative approaches are being introduced to overcome the limited accessibility of conventional PROTACs. Various innovative PROTAC technologies have been developed to enhance the selectivity for specific tissues and cells, including Ab-PROTAC, which uses antibodies to target cell membrane surface proteins for improved accessibility [53, 54]; pc-PROTAC, a UV-dependent PROTAC activated at specific locations using UV light [55]; CLIPTAC, which addresses cell permeability issues by linking E3 ligases and target proteins through intracellular chemical reactions [56, 57]; and Folate-PROTAC, which selectively delivers PROTACs to cancer cells with high FOLR1 expression [58]. These advances significantly expand the potential applications of PROTAC technology.
The main limitation of MGs is the lack of rational design strategies, making their discovery and development challenging. The effects of MGs are difficult to predict because they must induce PPIs to enable protein degradation or functional regulation. Furthermore, MGs cannot be applied to proteins not engaging in PPIs, limiting their target scope [23]. The rational development of MGs will require new platforms incorporating screening approaches focused on targets and effectors, along with chemical design strategies [59]. A combination of innovative biochemical assays (TR-FRET, AlphaScreen), cell-based screening methods (viability/toxicity analysis, reporter gene assays, NanoBRET), co-immunoprecipitation, proximity labeling techniques (BioID, TurboID, AirID), and computational approaches is crucial to screen and characterize MGs effectively [60–62]. New screening technologies and biochemical analysis methods for MG discovery are evolving. AI-based machine learning is expected to play a key role in predicting the MG mechanisms of action and optimizing drug design [63, 64].
CONCLUSION
TPD is an innovative approach that overcomes the limitations of SMIs by directly degrading target proteins, offering a novel therapeutic strategy for disease treatment. Among current TPD technologies, PROTACs and MGs are the most actively researched, showing promise as therapeutic agents for cancer, neurodegenerative diseases, and autoimmune disorders, with some candidates having entered clinical trials. Using E3 ligases to degrade the target proteins, TPD technologies can effectively target undruggable proteins that are difficult to modulate with conventional drugs. Nevertheless, challenges persist, such as PK limitations in PROTACs and target protein restrictions in MGs, necessitating further technological advancements and research efforts to overcome these limitations. The advances in TPD technology are driving the development of new drug discovery platforms that focus on identifying novel E3 ligases, optimizing drug design, and AI-driven drug development. These advances will enable TPD technology to establish itself as a more precise and effective therapeutic approach, playing a crucial role in treating various diseases, including refractory and difficult-to-treat conditions.