




INTRODUCTION
Cryptosporidium parvum is an obligate intracellular apicomplexan protozoon that infects the small intestine. Cryptosporidium is globally distributed and infects a wide range of hosts including livestock, poultry, and humans, and forms an important aspect of public health. Cryptosporidiosis, caused by C. parvum, disrupts the small intestinal villi and epithelial cells, leading to mucosal inflammation and a reduction in enzymatic activity [1]. This disruption results in compromised intestinal mucosal barriers, increased intestinal permeability, and the secretion of fluids and electrolytes, leading to dysfunctional digestion and absorption [2]. Consequently, the clinical symptoms of cryptosporidiosis vary from watery diarrhea, weight loss, poor growth performance, and decreased feed efficiency to life-threatening conditions, especially in immunodeficient and neonatal animals [3].
Caprine cryptosporidiosis mainly occurs in young animals (< 2 months old), and infected goat kids excrete a large number of oocysts (107–108 oocysts per gram) [4]. Cryptosporidiosis in adult goats is generally asymptomatic because of the host’s strong immune response; however, it exhibits 80%–100% morbidity and > 50% mortality in goat kids [5]. Therefore, controlling cryptosporidiosis is of great interest to the small ruminant industry. However, no effective drug or vaccine is currently available; instead, only sanitary management and supportive therapies such as rehydration, feed supplementation, and antidiarrheal agents have been employed.
Goats, as ruminants, possess a complex and distinctive digestive system that is well-suited for fermentation. Gut microorganisms present in ruminants play a pivotal role in the breakdown of plant fibers, significantly contributing to energy production [6]. These microorganisms generate up to 70% of the daily host enzyme requirements. Additionally, the gut microbiota actively stimulated villus growth in the rumen wall, demonstrating its impact on the overall digestive process [7]. Previous studies have shown that the structure and relative abundance of the caprine gut microbiota are affected by dietary changes, acquisition of colostrum and milk, the physiological environment of the gut, and administration of antibiotics [7-11].
Scientific interest in the association between parasitic infections and gut microbiota in animals is increasing because parasitic infections alter the host gut microbiota diversity and composition, leading to metabolic disorders [12-14]. Parasites infecting the gastrointestinal tract induce functional impairment of the gastrointestinal system, resulting in diarrhea [15]. Goat kids are particularly susceptible to parasitic infections and frequently experience diarrhea. Previous studies have shown that the composition of the ruminant gut microbiota is altered by the occurrence of diarrhea [9, 16]. However, limited research has been conducted on the impact of C. parvum, a diarrhea-causing pathogen, on the intestinal microbiota of goat kids.
Recently, metataxonomic analysis using high-throughput sequencing has been developed and implemented to study alterations in the diversity and composition of the gut microbiota in response to pathogenic infection [17]. This approach can deepen our knowledge of host–parasite interactions and provide alternative ways to control parasitic infections. Therefore, this study aimed to identify the effect of C. parvum infection on gut microbial composition and diversity in goat kids.
MATERIALS AND METHODS
In this study, 12 fecal samples were obtained from goat kids (< 1-month-old) on a single goat farm in Chungcheongbuk-do, Korea. Among the 12 fecal samples, seven were naturally infected with C. parvum showing diarrhea and five were from C. parvum-uninfected goat kids. Infection with C. parvum was verified by conventional polymerase chain reaction (PCR) as described in our previous study; however, the presence of other diarrhea causing pathogens was not verified [18]. All goats were raised in an intensive farm system for meat production and were of mixed breed. Information on the diet of goats was not collected.
Diarrheal feces were collected from the rectum using a sterile wooden stick, whereas nondiarrheal feces were collected immediately after defecation. All fecal samples were transported to College of Veterinary Medicine, Chungbuk National University, and stored at 4°C until further processing.
Genomic DNA was extracted from each fecal sample using the QIAamp® Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. DNA quality was assessed using a Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), and the DNA was stored at –20℃ until further processing.
The V3–V4 hypervariable region of the bacterial 16S rRNA gene was amplified using locus-specific primers (341F/805R), and sequencing was performed on the Illumina MiSeq platform (Illumina, San Diego, CA, USA) [19]. PCR was conducted in a total reaction volume of 25 μL containing 2.5 μL gDNA, forward and reverse primers (5 μL each; 1 μM), and KAPA HiFi Hotstart ReadyMix (12.5 μL; Kapa Biosystems, Boston, MA, USA) under following conditions: 95℃ initial denaturation for 3 min followed by 25 cycles of 95℃ for 30 s, 55℃ for 30 s and 72℃ for 30 s, and a final extension at 72℃ for 5 min. Nextera XT index adaptors (Illumina) were used for the index PCR. PCR products were purified with AMPure XP beads (Beckman Coulter, Brea, CA, USA). Amplicons were quantified using a Qubit dsDNA HS Quantification Assay (Thermo Fisher Scientific). DNA libraries were multiplexed and sequenced on an Illumina MiSeq system, according to the manufacturer’s instructions.
Raw paired-end Illumina sequences were processed using the dada2 pipeline (version 1.28.0) in R software (version 4.3.2) [20]. Briefly, the processes involved primers were trimmed and filtered using the “filterAndTrim” function. Reads exceeding 4 (forward) and 6 (reverse) expected errors based on quality scores were filtered out, and truncation was performed after 280 (forward) and 260 (reverse) base pairs. The dada2 algorithm was applied to infer the exact amplicon sequence variants (ASVs) from the processed sequences, while filtering chimeras. For taxonomic assignment, a naïve Bayesian classifier method was used based on the SILVA reference database (version 138; https://www.arb-silva.de) with a minimum bootstrap confidence of 50. An ASV table was constructed by recording the number of times that each ASV was observed in each sample.
The ASV table, sample data, and taxonomy table were imported into the R environment as “phyloseq” objects (version 1.44.0) [21]. Additional sample data categories were added to the phyloseq sample data table. ASVs without assigned phyla were filtered and agglomerated at the species level. Alpha and beta diversity metrics were calculated on the filtered ASV table using Phyloseq’s estimate_richness and distance functions. Taxonomy summaries and microbial composition bar plots were generated at both phylum and class levels.
RESULTS
As a raw data, 1,873,294 pairs of readings (mean: 156,107.83; S.D.: 11,472.82) were obtained. After processing the raw sequences, 1,274,317 (mean: 106,193.08; S.D.: 6,865.77) high-quality read pairs were obtained. In total, 256 ASVs were identified based on the SILVA reference database. The number of ASVs according to the samples varied from 85 to 126 (mean: 108.17; S.D.: 12.14).
Alpha diversity indices were used to analyze species richness. The Observed, InvSimpson, Chao1, and abundance-based coverage estimators diversity indices showed no significant differences in the gut microbial biodiversity between C. parvum-infected and uninfected goat kids (p>0.05). However, the Shannon diversity index was statistically significant (p<0.05; Fig. 1A).
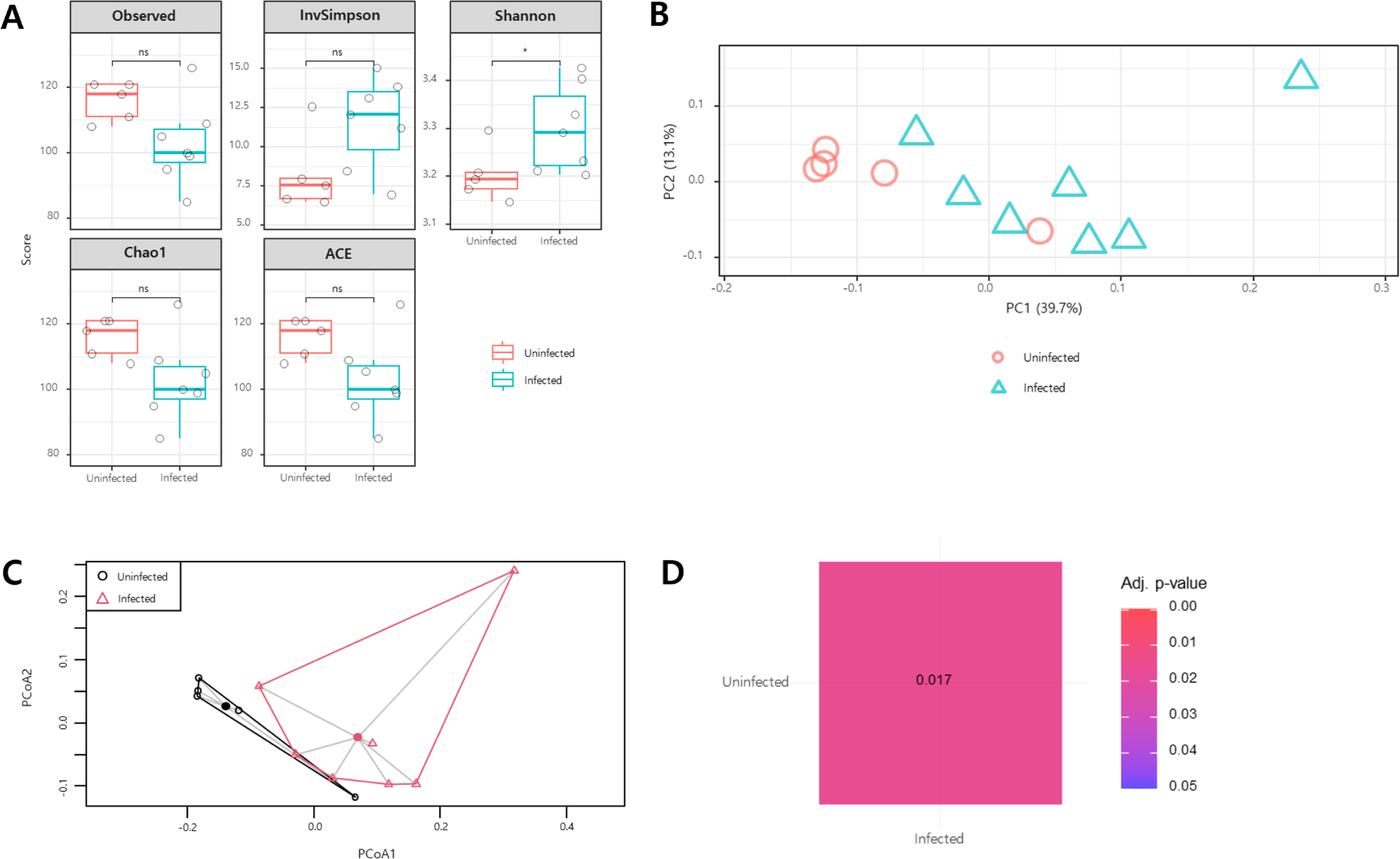
Beta-diversity analysis was conducted to assess the microbial community composition. In principal coordinate analysis using the Bray–Curtis distance and Jaccard distance methods, samples from the same group were clustered together (Fig. 1B and C). The results of the pairwise permutational multivariate analysis of variance revealed differences in the microbial composition between C. parvum-infected and uninfected animals (p<0.05; Fig. 1D).
The taxonomic structure of the gut microbial composition in C. parvum-infected and uninfected goat kids was assessed. At the phylum level, Bacteroidetes and Firmicutes were predominant in both groups, followed by Proteobacteria and Actinobacteria (Fig. 2A). No significant difference in the Bacteroidetes/Firmicutes ratio was observed between the two groups (p>0.05; Fig. 3A).
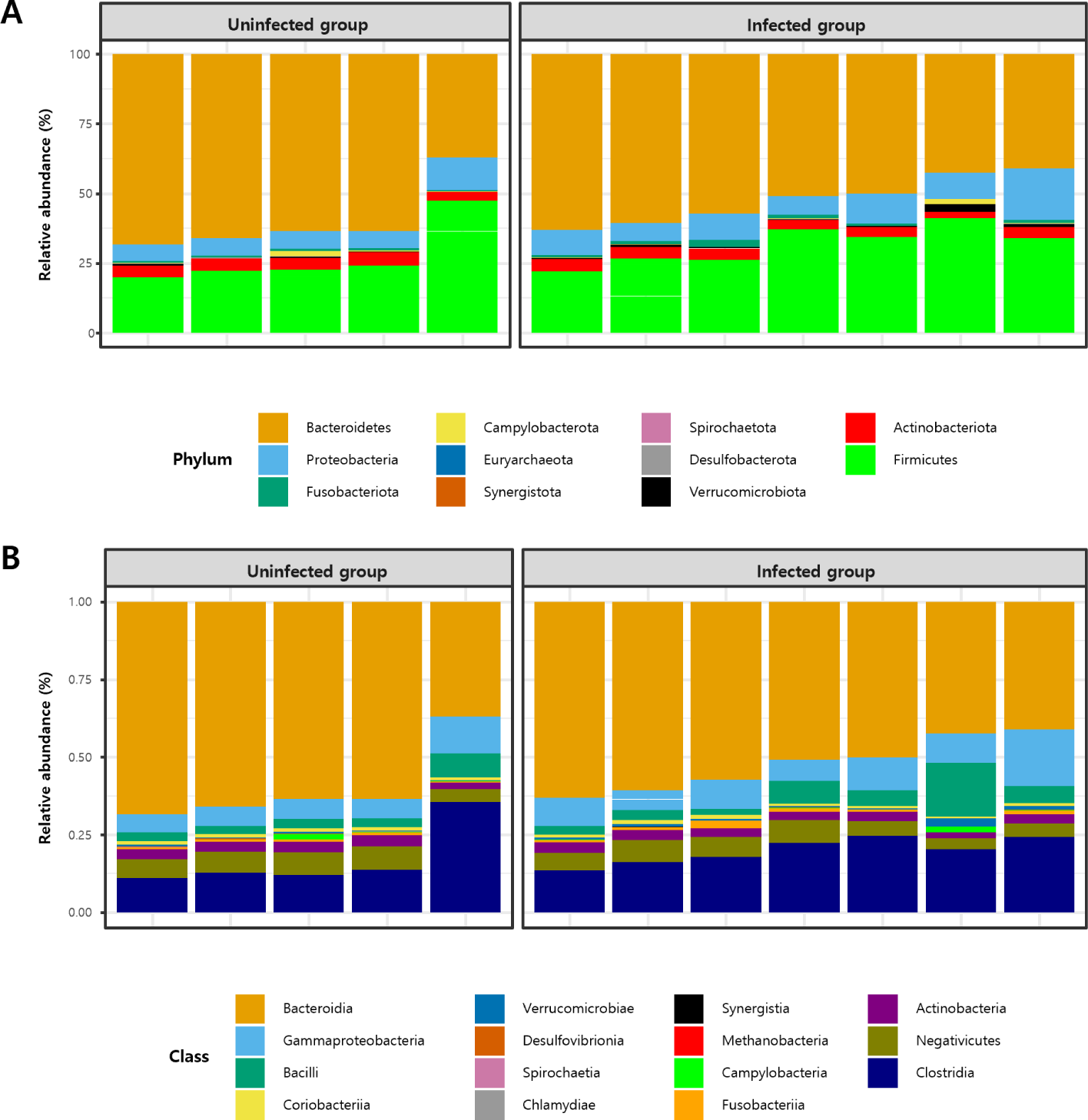
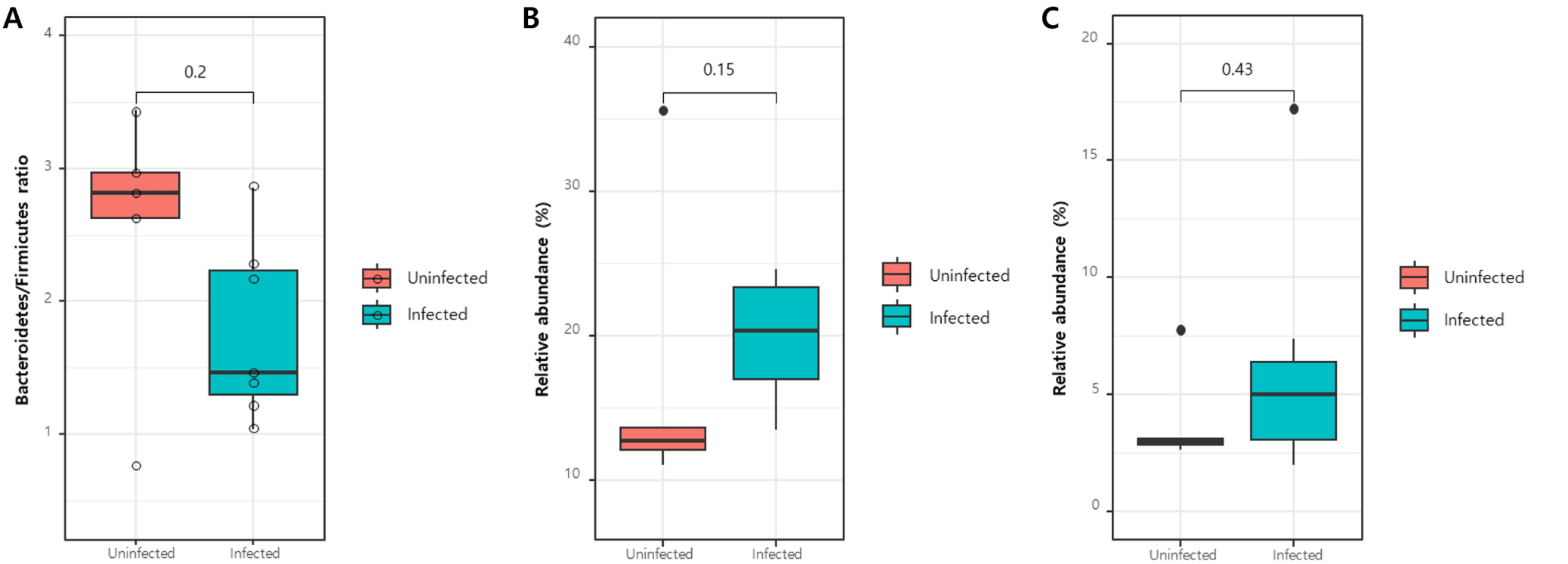
At the class level, Bacteroidia, Clostridia, Gammaproteobacteria, Bacilli, and Negativicutes were predominant (Fig. 2B). The relative abundances of Clostridia and Bacilli, known for their digestive roles in ruminants and belonging to the phylum Firmicutes, were compared; however, no significant differences were observed in either case (p>0.05; Fig. 3B and C).
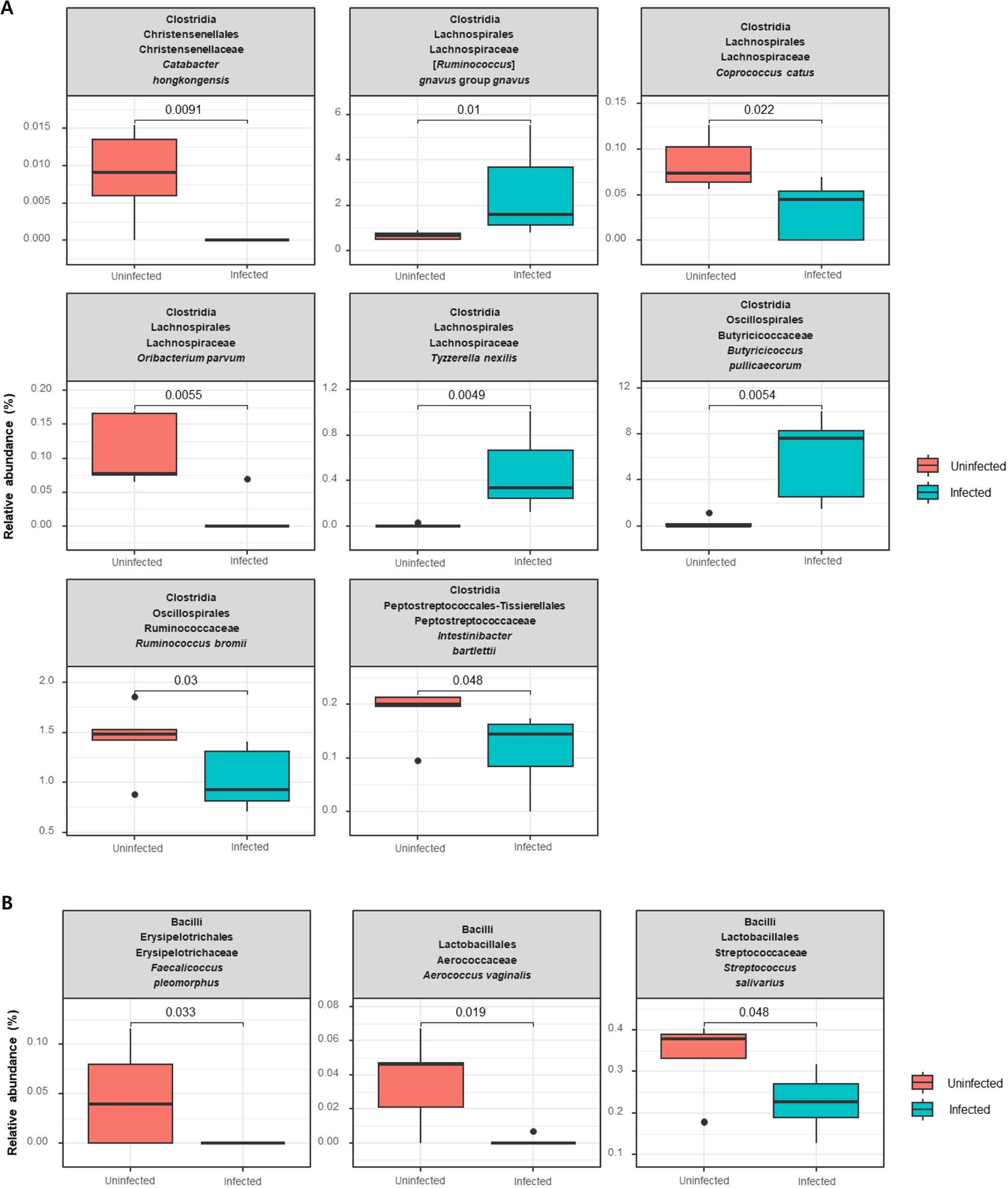
At the species level, within the class Clostridia, Catabacter honkongensis, Coprococcus catus, Oribacterium parvum, Ruminococcus bromil, and Intestinibacter bartlettii were negatively associated with C. parvum infection, whereas Ruminococcus gnavus, Tyzzerella nexills, and Butyricicoccus pullicaecorum were positively associated (p<0.05; Fig. 4A). In addition, at the species level, within the class Bacilli, Faecalicoccus pleomorphus, Aerococcus vaginalis, and Streptococcus salivarius showed decreased relative abundance in the C. parvum-infected group (p<0.05; Fig. 4B).
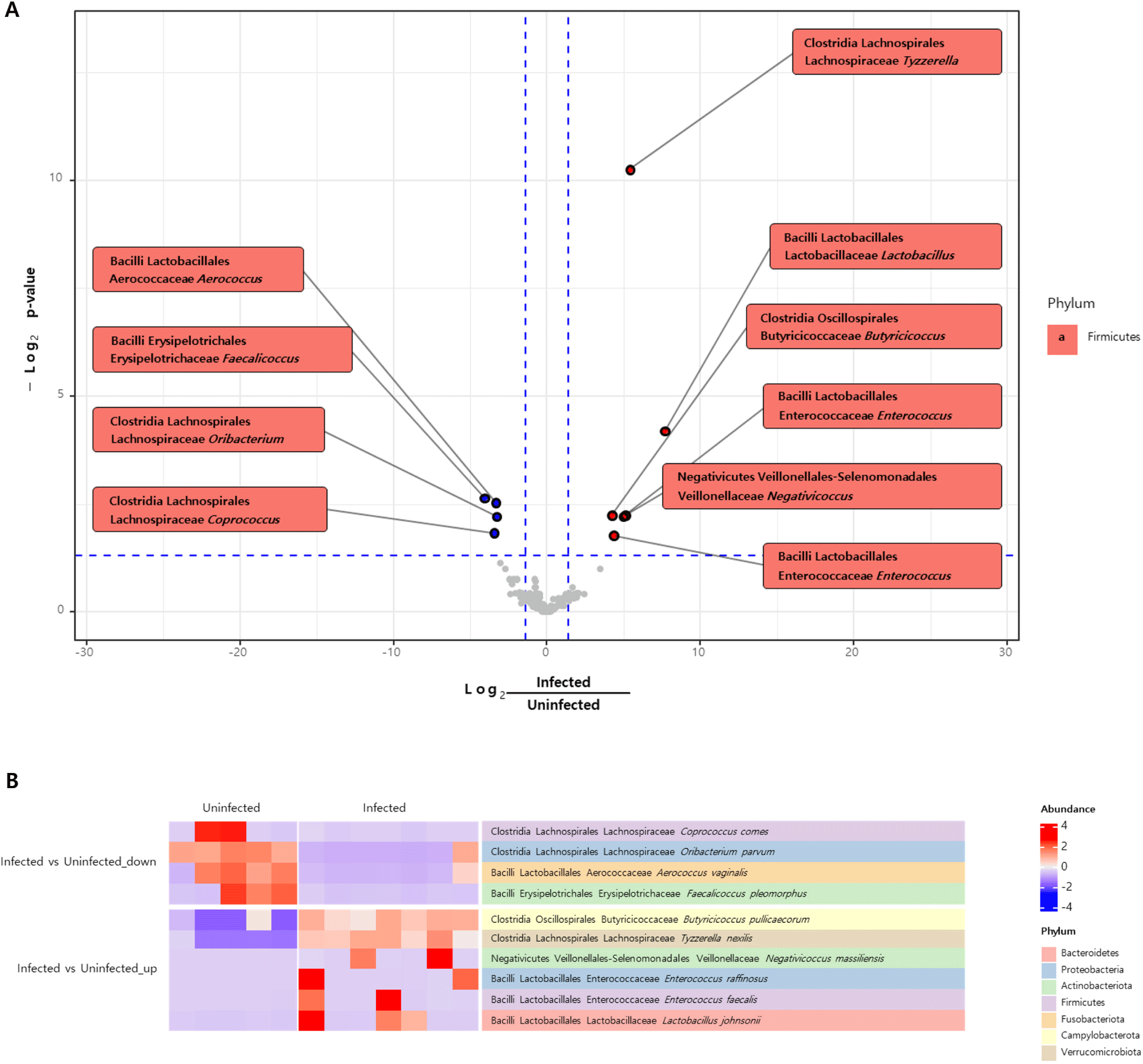
Differential abundance analysis was conducted to identify the significant differences in the abundance of taxa between the two groups. A volcano plot revealed elevated levels of Tyzzerella, Lactobacillus, Butyricicoccus, Enterococcus, and Negativicoccus whereas Aerococcus, Faecalicoccus, Oribacterium, and Coprococcus were less abundant in the C. parvum-infected group than in the uninfected group (Fig. 5A). At the species level, the heatmap indicated a notable increase in the abundance of T. nexillis, Lactobacillus johnsonii, B. pullicaecorum, Enterococcus raffinosus, Enterococcus faecalis, and Negativicoccus massiliensis, and a decreased abundance of A. vaginalis, F. pleomorphus, O. parvum, and Coprococcus comes (Fig. 5B).
DISCUSSION
Several studies have reported alterations in the gut microbiota in response to Cryptosporidium infection in various animals [22-27]; however, there are few studies involving goats. In this study, the differences in microbial diversity between C. parvum-infected and uninfected goat kids were analyzed. Alpha diversity, which measures the number of species and their relative abundance, showed no significant differences except for the Shannon index. However, beta diversity, which assesses the differences in species composition between groups, indicated significant shifts in the microbial community composition due to C. parvum infection (p<0.05). This means that C. parvum-infected and uninfected goat kids harbored different specific species and their abundances. These findings demonstrate the impact of C. parvum infection on altering the gut microbial taxonomic composition and diversity.
A previous study showed that C. parvum infection dramatically alters gut microbial diversity in goat kids at 5 days post infection (dpi), whereas the microbial composition and diversity of the C. parvum-infected group are not significantly different from those of the control group at 15 dpi [28]. In the present study, PERMANOVA analysis showed a significant difference in microbial composition between the two groups (p<0.05), suggesting that the goats were in the early stages of C. parvum infection. PCoA is used to visualize the diversity and composition of microbial communities, where samples with similar microbial compositions clustered together on the graph. In this study, using the Bray-Curtis distance and Jaccard distance methods to represent dissimilarities, the C. parvum-infected group appeared distinct from the uninfected group, indicating the impact of this parasite on microbial communities in goat kids.
In the digestive system of ruminants, microbiota plays a crucial role in generating host enzymes, highlighting the importance of microbial diversity and abundance [14]. Firmicutes and Bacteroidetes are predominant in goat kids regardless of age or health status [9]. Firmicutes degrade fiber and cellulose in ruminants and produce short-chain fatty acids that regulate immune responses. Bacteroidetes are involved in the breakdown of carbohydrates and proteins and contribute to the development of the gastrointestinal immune system [14]. In a previous study, mice experimentally infected with C. parvum showed an elevated Bacteroidetes/Firmicutes ratio compared with the uninfected group [4]. Similar findings have been observed in pigs infected with Balantioides coli and mice infected with Giardia [17, 29], suggesting a common role for Bacteroidetes and Firmicutes in the gut microbiota. In this study, these two bacterial phyla were predominant in both groups, which is consistent with the results of previous studies; however, no significant difference in the ratio between these two groups was noted.
Analysis of microbiota diversity, conducted using a differential abundance test, revealed significant alterations in the microbial composition. Clostridia and Bacilli belonging to the phylum Firmicutes are involved in the digestive processes of ruminant animals. However, the abundance of Clostridia and Bacilli in C. parvum-infected goat kids was not significantly different from that in uninfected goat kids. As only a limited number of species within both classes exhibited significant alterations, further studies at the class level are required.
The genus Lactobacillus, which belongs to the class Bacilli, produces lactic acid [14]. Lactobacillus is often associated with healthy gut and is commonly used as an ingredient in probiotics. In this study, C. parvum-infected group showed statistically higher abundance of Lactobacillus which is different from our expectation. Previous studies on goat kids’ gut microbiome did not show a difference according to C. parvum infection [9, 28]. It is worth noting that a previous study suggested that C. parvum infection may be a cause of the development of secondary lactose intolerance [30]. Due to the limitation of the tested animal numbers in this study, we cannot conclude clear association between C. parvum infection and Lactobacillus, further studies are required.
In humans, Tyzzerella abundance is elevated in individuals with cardiovascular diseases. Furthermore, a pronounced increase in Tyzzerella is evident in individuals who follow unhealthy dietary patterns or are afflicted with Crohn’s disease. A previous study revealed that administering a probiotic supplement of bifidobacteria to mice resulted in a reduction in Tyzzerella levels [31]. In the present study, a significant increase in this genus was observed in the C. parvum-infected group, suggesting an association between Tyzzerella and C. parvum infection.
Enterococcus spp. are found in the intestinal flora of humans and E. faecalis is a common species known for its immunomodulatory role in both humans and animals [32]. Some species are used as probiotics; however, enterococci can also serve as pathogens. Previous studies have demonstrated the abundance of E. faecalis and E. raffinosus in patients with ulcerative colitis (UC) or inflammatory bowel disease (IBD) [33, 34]. The effects and pathogenesis of the increased presence of enterococci are not fully understood [35]; however, an association between these bacteria in the gut microbiota and UC or IBD has been observed [33, 34]. In the present study, the genus Enterococcus showed a significant increase in the group infected with C. parvum; this trend was also observed for E. faecalis and E. raffinosus. The precise effect of these bacteria remains unclear; nevertheless, they appear to be linked to alterations in the intestinal wall. Additionally, these bacteria notably exhibit antibiotic resistance, emphasizing the need for further research to better understand their roles and implications.
The gut microbiota is influenced by different internal and external factors such as age, diet, and antibiotic usage [7, 9]. The results of this study showed that infection with C. parvum and diarrhea affected the gut microbiota; however, some results were inconsistent with those of previous studies, such as the Bacteroidetes/Firmicutes ratio and microbial composition. As the goats included in this study were reared on a farm located in the environment, all samples were collected from a single farm to minimize external environmental factors. However, the farm differs from well-controlled experimental conditions, and various factors, including infection intensity (quantity of Cryptosporidium), period of diarrhea, dietary variations, and age, may influence the gut microbiota [14, 28].
This study examined the diversity and compositional changes in the gut microbiota of goat kids after C. parvum infection. The findings of this study showed that C. parvum infection, associated with diarrhea in neonatal goats, modulates the caprine intestinal microbiota. This research provides valuable insights into the interaction between C. parvum infection and the gut microbiota, thereby advancing our understanding of the pathogenesis of diarrhea associated with this parasite.