




INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic inflammatory disease of the small and large intestines. Ulcerative colitis and Crohn's disease are the two main forms of IBD. Several experimental results have shown that the dysregulation of CD4+ T helper cell (Th cell) function can lead to intestinal mucosal inflammation. Experimental evidence from mouse models has shown that the adoptive transfer of naïve CD4+ T cells without residual activated/memory/Treg cells into immunodeficient recipient mice initiates IBD development [1].
T helper (Th) play important roles in the adaptive immune response by coordinating the expansion and regulation of CD8+ T cells, macrophages, and B cells, and recruiting innate immune cells to the site of inflammation. Upon antigenic stimulation, naïve CD4+ Th cells differentiate into specialized effector subsets, namely T helper 1 (Th1), T helper 2 (Th2), T helper 17 (Th17), T follicular helper (Tfh), and regulatory T (Treg) cells, with distinct patterns of cytokine production and effector functions [2]. Th1 cells are differentiated upon stimulation with interleukin (IL)-12 via signal transducer and activator of transcription 4 (STAT4) signaling and upon interferon (IFN)-γ stimulation via STAT1, which induces expression of transcription factor T-box 21 (T-bet) and of IFN-γ. Th2 cells are induced by IL-4 via STAT6 signaling, which promotes the expression of the transcription factors GATA3, IL-4, IL-5, and IL-13. Classically, Th1- and Th2-mediated immune responses have been linked to IBD pathogenesis [3]. However, numerous studies have shown that an imbalance between Th17 and Treg cells contributes significantly to IBD development [4–6]. Owing to their significance in the gut mucosal immune response and contribution to autoimmune disorders, Th17 cells have received increasing attention in recent years. In this review, we discuss the role of Th17 cells in IBD pathogenesis and their therapeutic potential.
TH17/TREG BALANCE IN THE INTESTINE
Th17 cells are exclusively abundant in the small and large intestinal lamina propria (LP), where they contribute to the mutualistic link between the host and microbiota. Th17 cells have protective roles against bacterial and fungal infections that invade at intestinal barrier. However, uncontrolled Th17 cells activity has been linked to several autoimmune diseases. Compared to Th17 cells, Treg cells play a critical role in maintaining immune tolerance and suppressing inflammation to maintain tissue homeostasis. However, when this Th17/Treg balance is disrupted, mice become vulnerable to various infections and autoimmune diseases. Therefore, the Th17/Treg cell balance in the intestine is important to maintain gut integrity against bacterial and fungal infection, while preventing autoimmune disease, such as IBD. Abundant Th17 cells are commonly seen in patients with IBD and mouse models of colitis. These cells primarily contribute to the development of IBD by secreting cytokines, such as IL-17, IL-22, and IL-26. This review focuses on Th17 cell differentiation and the involvement of Th17 cells in IBD development and progression.
TH17 CELLS IN INTESTINAL HOMEOSTASIS
IL-17 and IL-22 secreted by Th17 cells induce antimicrobial peptide production to maintain the barrier integrity under homeostatic conditions [7]. IL-17 activates stromal and myeloid cells to produce G-CSF, which induces neutrophil production in the bone marrow and produces chemokines that recruit neutrophils to the gut. IL-22 induces goblet cell hyperplasia and helminth expulsion during intestinal infection. Additionally, intestinal epithelial cells (IECs) express IL-23 receptor (IL-23R) under homeostatic conditions and may respond to IL-23 stimulation by producing protective IL-22, which supports the intestinal barrier function. Moreover, CCL20 a chemokine produced by activated epithelial cells and Th17 cells, increases the recruitment of CCR6-expressing Th17 cells to the site of infection. Consequently, genetic defects in IL-17a and IL-17f in mice increase their susceptibility to opportunistic infections of mucosal bacteria, such as Staphylococcus aureus and Citrobacter rodentium [8]. Therefore, Th17 cells are critical for defense against pathogenic extracellular bacteria and fungi.
PATHOGENIC TH17 CELLS IN THE PATHOGENESIS OF INFLAMMATORY BOWEL DISEASE
Besides the regulation of intestinal flora, Th17 cells may also exhibit pathogenic features, particularly following their stimulation with IL-23, IL-1β, transforming growth factor (TGF)β3, and serum amyloid A antigen (SAA). Pathogenic or non-pathogenic differentiation of Th17 cells is regulated by distinct cytokines (Fig. 1). For example, in vitro polarization of naïve CD4+ T cells by IL-6 and TGFβ1 stimulation generates non-pathogenic Th17 cells that produce IL-17 and IL-10, however, induce a little tissue inflammation, whereas IL-6, TGFβ1, and IL-23, or IL-1β, IL-6, and IL-23 can induce pathogenic Th17 cells that contribute to tissue inflammation [9–11]. IL-6 along with SAAs or TGFβ3 can also induce pathogenic Th17 cells [12] (Fig. 1).
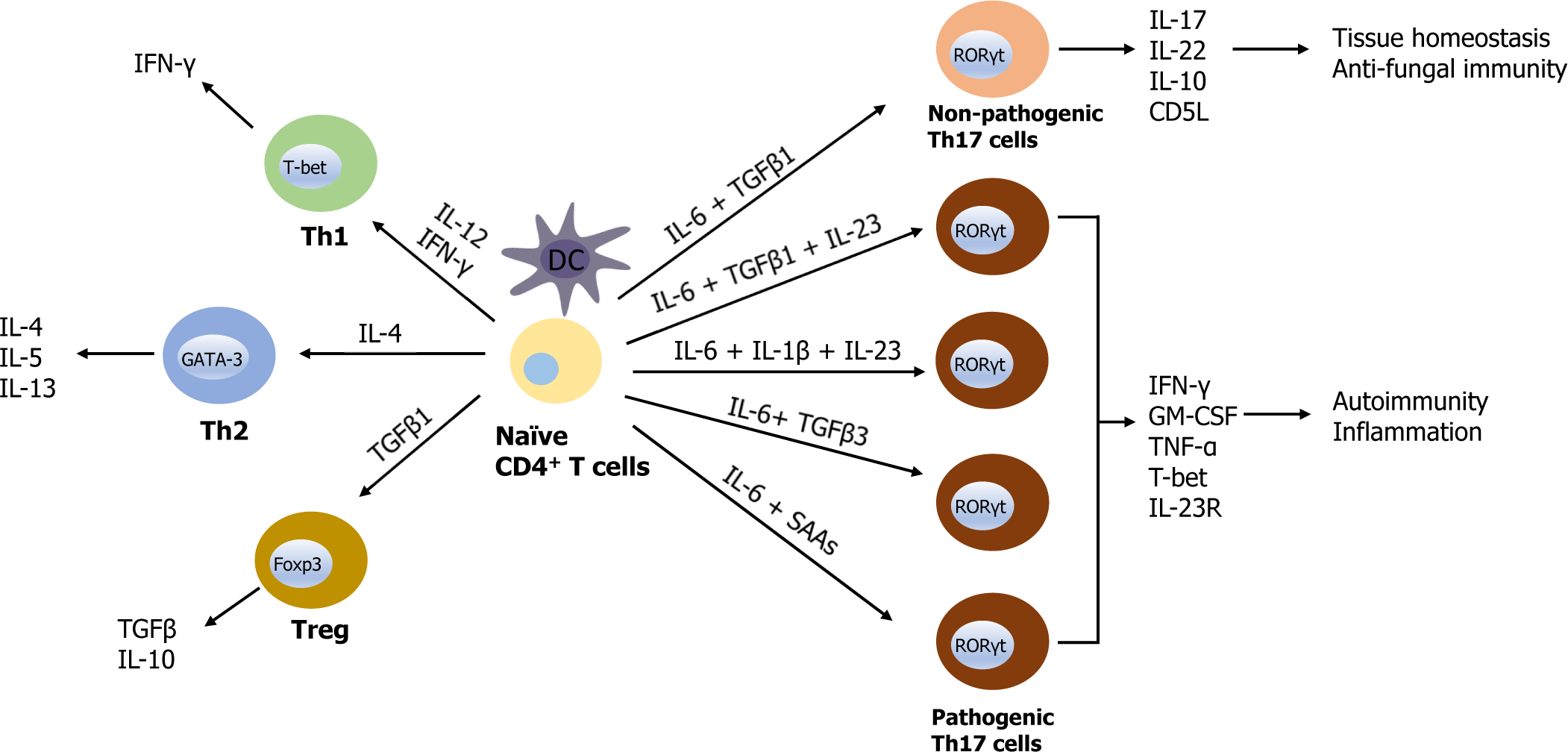
IL-10 producing non-pathogenic Th17 cells promote tissue homeostasis and protection [13], whereas pathogenic Th17 cells secrete proinflammatory cytokines such as IFN-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), and tumor necrosis factor (TNF)-α, exacerbating autoimmune disorders [14]. IL-23 drives intestinal Th17 proliferation and enhances the development of pathogenic T cells that secrete proinflammatory cytokines such as IFN-γ [15], while IL-23R signaling negatively regulates the survival of intestinal Treg cells [16, 17]. Although pathogenic and non-pathogenic Th17 cells are dependent on the retinoic acid receptor-related orphan receptor γt (RORγt) for their differentiation, the switch between non-pathogenic and pathogenic Th17 cells remains an unelucidated.
Th17 cells transdifferentiate into Th1 and Treg cells. IL-12 and IL-23 can both induce the conversion from Th17 cells into Th1 cells by altering cytokine secretion from IL-17 to INF-γ in a STAT4- and T-bet-dependent manner, which is required for the pathogenesis of colitis [15]. Notably, IL-23 drives intestinal inflammation, promotes intestinal Th17 cell accumulation, and enhances the emergence of IL-17+IFN- γ+ colitogenic T cells in the gut [15, 17]. Moreover, Th17 cells can promotes colitis-associated intestinal fibrosis, which can result in internal strictures, structural distortion, and loss of function. IL-17A inhibits migratory capacity and promotes the production of the extracellular matrix collagen of myofibroblasts [18]. IL-6 and IL-21 stimulate the expression of amphiregulin expression in Th17 cells [19]. Amphiregulin increases proliferation and collagen expression in intestinal myofibroblast, which leads to more severe intestinal fibrosis [19]. Treg cell therapy has been shown to be beneficial in several IBD models. In the presence of Symbiotic segmental filamentous bacteria (SFB), some intestinal Th17 cells lose IL-17A expression and acquire IL-10 expression [20]. Circulating IL-17 producing FOXP3+ CD4+ T cells are increased, while the suppressive activity of Tregs is significantly reduced in patients with IBD compared with that in healthy control subjects [21]. These results suggested plasticity in the transdifferentiation of Th17 and Treg cells. Thus, Th17 cell plasticity plays an important role in regulating intestinal immune responses in patients with IBD.
TH17 CELL DIFFERENTIATION AND REGULATION
Th17 cells were first discovered in 2005 as a Th cell lineage that is independent of Th1- and Th2-related transcription factors (T-bet, STAT1, STAT4, and STAT6) [22]. Th17 cell differentiation can be divided into three stages: induction (driven by TGFβ and IL-6/IL-21), amplification (triggered by IL-21), and stabilization (maintained by IL-23 and IL-1β). Naïve CD4+ T cells can be differentiated into Th17 cells in the presence of TGFβ, which in turn drive SMAD signaling, and IL-6 and IL-21, which activate the transcription factor STAT3 [23]. Phosphorylated STAT3 interacts with the IL-17 promoter and activates the master transcription factors of Th17 cells, RORγt and RORα, which promotes expression of the IL-23R on the surface of Th17 cells [24]. Thus, differentiation and activation of Th17 cells is stringently governed by various cytokines, multiple transcription factors (AP-1, JunB, aryl hydrocarbon receptor [Ahr], IRF4, BATF, c-Maf, PLZ, PPARγt, and EGR-2), and environmental factors such as gut microbes and their metabolites.
CYTOKINES MODULATING DIFFERENTIATION AND ACTIVATION OF TH17 CELLS
IL-6 is a pleiotropic cytokine produced by several cell types, including macrophages, dendritic cells (DCs), monocytes, endothelial cells, and IECs. Although various hematopoietic and non-hematopoietic cells can produce IL-6, Korn et al. reported that SIRPa+ IRF4-expressing DCs, equivalent to murine intestinal CD103+CD11b+ DCs, are crucial for priming pathogenic Th17 cells by transplanting IL-6 into T cells [25]. IL-6 stimulates Th17 cell development by upregulating RORγt through the JAK2-STAT3 signaling pathway [26]. IL-6 signaling is critical for maintaining the Th17/Treg balance, because IL-6 inhibits TGFβ-induced Treg differentiation [11]. Phosphorylated STAT3 can inhibit TGFβ-induced expression of FOXP3, which encodes a transcription factor that binds and antagonizes the function of RORγt, and thereby inhibits the generation of Treg cells. Hyperactivation of STAT3 in naïve CD4+ T cells promotes Th17 cell differentiation, whereas ablation of STAT3 impairs Th17 cell differentiation and skews towards differentiation into FOXP3+ Treg cells [24, 27]. IL-6 triggers the expression of IL-21, which amplifies the autocrine loop and further induces the expression of IL-21 and surface IL-23R on naïve CD4+ T cells [26]. In addition, in IBD, macrophages release IL-6, which facilitates resistance to apoptosis and accumulation of aberrant T-cell in the intestinal mucosa, and promotes the persistence of pathogenic Th17 cells [28].
TGFβ is also a pleiotropic cytokine and can be produced by many types of cells, such as epithelial cells, DCs, T cells, and fibroblasts. TGFβ is abundant in the intestinal cells because its production is upregulated by various factors, such as bacteria, viruses, and cytokines. TGFβ is required for differentiation of Th17 and Treg cell and can induce both RORγt and FOXP3 expression via SMAD signaling. At high concentrations, TGFβ inhibits IL-23R expression and favors FOXP3+ Treg differentiation by antagonizing RORγt, whereas low TGFβ levels combine with IL-6 and IL-21 to enhance the expression of IL-23R and promote Th17 cell differentiation [11]. In Th17 cells, TGFβ promotes IL-22 production via Ahr induction and PI3K signaling [29]. IL-23 is a proinflammatory cytokine with a heterodimeric structure composed of the p19 and p40 subunits. The IL-23 p40 subunit is shared by the IL-12 p40 subunit [30]. IL-23 signals via the IL-12Rβ1 subunit (shared with IL-12-p40) and its unique IL-23Rα subunit.
IL-23R in turn stimulates JAK2 and TYK2 and activates STAT3, promoting transcription of IL-23R and RORC (which encodes RORγt) [31]. IL-12 is a heterodimer cytokine composed of the IL-12 p40 subunit and the IL-12 p35 subunit, and it signals through IL-12Rβ1 and IL-12Rβ2 [30]. The difference in IL-23- and IL-12-dependent signaling is partly due to the preferential activation of STAT3 by IL-23, and STAT4 by IL-12. IL-23 is mainly secreted by DCs, tissue-resident macrophages, and neutrophils upon non-self (microbial products such as lipopolysaccharide and peptidoglycans) and self-signals (prostaglandin E2 and adenosine 5’-triphosphate [ATP]) upon risk or injury [32]. The production of IL-23 can be further increased by CD40–CD40L interactions, which drive a positive feedback loop in DC activation [33]. IL-23 is not a Th17 cell-differentiating factor instead acts on previously differentiated Th17 cells, stabilizing their pathogenic function, in the absence of IL-23R in naïve CD4+ T cells [10, 34]. IL-23 can promote Th17 cell proliferation and secretion of proinflammatory cytokines, such as IFN-γ, GM-CSF, and TNF-α [17, 30, 34]. IL-23 promotes intestinal inflammation and exacerbates IBD progression [35, 36]. Results of a meta-analysis suggest that genetic polymorphisms in IL-23R are significantly associated with susceptibility to IBD [37]. In addition, activation of IL-23R signaling impairs the stability and function of intestinal Tregs, indicating that IL-23 affects the Th17/Treg cell balance in IBD [16].
IL-1β was first described as a lymphocyte-activating factor that stimulates T lymphocytes. IL-1β performs a range of proinflammatory activities. It is primarily produced by activated macrophages, monocytes, T-cells, NK cells, and endothelial cells [38]. IL-1 and IL-23 can be secreted by DCs upon stimulation with microbes, such as the bacterial NOD2-ligand muramyl dipeptide [39]. IL-1 is expressed as an inactive precursor, which must be proteolytically cleaved by the enzyme caspase-1 to become the active IL-1β cytokine. The key mediator of IL-1 cleavage in the intestine is the NOD-like receptor protein 3 (NLRP3) inflammasome, which activates caspase-1. Under intestinal inflammatory conditions, the NLRP3 inflammasome-expressing intestinal macrophages and DCs are activated and further secrete IL-1β [40]. IL-1β increases the intensity and duration of pSTAT3 signaling by inhibiting SOCS3, a feedback inhibitor of JAK2/STAT3 signaling [41]. IL-1β controls transcription factors, including IRF4, RORγt, BATF, and NFKBZ [42]. Thus, IL-1β enhances Th17 cell differentiation and inhibits Treg cell differentiation by suppressing TGFβ-induced FOXP3 expression in naive CD4+ T cells. IL-1β synergizes with IL-6 and IL-23 in the intestinal LP to generate pathogenic Th17 cells that produce IFN-γ, independently of TGFβ1 [9, 43]. IL-1 signaling is essential for driving the development of colitis by promoting the accumulation and survival of Th17 cells in the intestine [44].
IL-21 is a pleiotropic cytokine primarily produced by Th17, NKT, and T follicular helper cells. IL-21R is expressed in various cells, including T cells, NK cells, DCs, and NKT cells. IL-21 secreted by the Th17 cells stabilizes and expands the Th17 lineage in an autocrine manner. IL-21 is produced by Th17 cells, and upregulates IL-17 production and expression of RORγt in a STAT3 dependent manner [45]. IL-21 and TGFβ1 together can also induce Th17 cell differentiation in the absence of IL-6, and IL-21R-deficient T cells fail to promote Th17 cell differentiation [45]. IL-21 promotes IL-23R expression in naïve CD4+ T cells, which increases cellular responsiveness to IL-23 [26]. Several studies have focused on the pro-inflammatory effects of IL-21 signaling in IBD [46, 47], whereas some studies reveal that IL-21 signaling suppresses intestinal inflammation [48]. The precise involvement of IL-21 signaling in IBD warrants further investigation.
DENDRITIC CELLS AND TH17 CELLS
DCs are professional antigen-presenting cells that regulate T-cell tolerance and priming. In the intestine, DCs continuously encounter harmless food antigens and commensal microbes that maintain homeostasis. Under steady-state conditions, DCs can induce T-cell tolerance to harmless antigens to avoid undesirable immune responses. In other contexts, T-cell priming by DCs, which induce proinflammatory responses against pathogens and/or infected cells.
The importance of DCs in the pathogenesis of IBD has been demonstrated previously. In animals with dextran sulfate sodium (DSS)-induced colitis, transfer of bone marrow-derived DCs worsens inflammation, whereas depletion of bone marrow-derived DCs reduces inflammation [49]. The administration of an agonistic anti-CD40 antibody to T- and B-cell-deficient mice was adequate to activate DCs and induce IL-23-dependent intestinal inflammation [50]. In CD, LP DCs are activated and produce high levels of IL-12 and IL-6 [51]. Production of proinflammatory cytokines by DCs is correlated with disease and gut microbiota composition [52]. In the mesenteric lymph nodes (MLNs) of patients with CD, DCs produce high levels of IL-23, IL-17, and IFN-γ upon bacterial stimulation, and their CD4+ T cells secrete increased levels of IL-17 and IFN-γ [53].
Intestinal DCs are heterogeneous, however, they can be classified into three major subsets: CD103+CD11b+ DCs, CD103+CD11b– DCs, and CD103–CD11b+ DCs [54]. These three DC subsets upregulate CCR7 levels and migrate to the MLNs, and instruct naïve CD4+ T cells to respond to intestinal antigens and drive Th17 cell differentiation [55]. In particular, IRF4 transcription factor-expressing intestinal DC subsets (CD103+CD11b+ and CD103–CD11b+) promote Th17 cell differentiation in MLNs [56]. Mice lacking IRF4-dependent DCs showed a reduced number of Th17 cells in their MLNs and intestines [57]. These studies show that IRF4 expressing DCs promote Th17 cell development in MLNs, primarily through the secretion of IL-6 [25, 57].
Innate immune signaling is important for regulating Th17/Treg immune balance in the intestinal tract. DC activation by several Toll-like receptor (TLR) ligands, including TLR2, TLR3, and TLR9, induces secretion of IL-6, IL-1β, and IL-23, which can promote differentiation of Th17 cells and gut inflammation [57, 58]. DCs that phagocytose apoptotic cells in the absence of microbial signals secrete TGFβ, which can promote Treg cell development. However, phagocytosis of infected apoptotic cells triggers the combined expression of IL-6 and TGFβ in a T-cell receptor-dependent manner, which alternatively promotes Th17 cell development [59]. These results suggest that DCs influence the development and activity of Th17 cells.
MICROBIOTA AND DIET-DERIVED SIGNAL-DEPENDENT REGULATION OF TH17 CELLS
Microbiota influences the development and homeostasis of the immune system and is associated with IBD. Germ-free (GF) mice show markedly reduced numbers of LP Th17 cells [7]. The colonization of GF mice with different complex microbiota or their metabolites can increase the number of Th17 cells in the intestine [7]. GF- and antibiotic-treated mice showed attenuated intestinal inflammation; however, weakened intestinal barrier function in DSS-induced colitis [60]. The introduction of microbiota from humans with IBD into GF mice increased the number of Th17 and Th2 cells and decreased the number of Treg cells, whereas the transfer of microbiota from healthy donors increased the number of Treg cells [4]. Colonization of GF mice with human microbiota in IBD also increases colonic inflammation and exacerbates disease severity in a T-cell transfer-induced colitis model [4]. Recent clinical trials have shown that IBD can be treated using fecal microbiota transplantation (FMT) [61].
Adhesion of microbes to IECs is crucial for Th17 cell development and effector function, thus maintaining the mucosal barrier function. SFB are among the most potent and well-characterized commensals that induce antigen-specific Th17 cell differentiation in the terminal ileum of the small intestine [62]. SFB adhere tightly to the IECs of the ileum. SFB upregulate reactive oxygen species and transfer SFB-associated proteins into IECs via microbial adhesion-triggered endocytosis, thus upregulating IL-1β and IL-23 production from CX3CR1-expressing macrophages [63]. CX3CR1-expressing macrophage-derived IL-23 and IL-1β activate group 3 innate lymphoid cells (ILC3s) to produce IL-22 [64], which stimulates the secretion of SAA 1 and 2 [65]. The co-culture of SFB and epithelial cell lines can also increase SAA expression in vitro [66]. SAAs stimulate DCs to produce IL-6, TGFβ, IL-23, and IL-1β, resulting in Th17 cell development and survival [65]. These steady-state Th17 cells are required for protection against the intestinal pathogenic bacteria [67, 68]. Notably, SAAs are significantly upregulated in patients with IBD [69]. SAAs can directly induce the differentiation and pathogenicity of colitogenic Th17 cells, thereby exacerbating colonic inflammation in IBD [12]. Moreover, 20 bacterial strains isolated from the feces of UC patients showed epithelial cell-adhesive characteristics and induced Th17 cells in the mouse colon [68].
Multiple environmental factors contribute to intestinal Th17 cell activation. For example, ATP derived from symbiotic bacteria stimulates CD70hiCD11clo LP cells to produce TGFβ, IL-6, and IL-23, thus promoting Th17 cell differentiation [70]. A high-salt diet increased the risk of colitis by inducing a pathogenic Th17 response in mice [71]. High-salt conditions stimulate p38/MAPK signaling involving NFAT and SGK1 during cytokine-induced Th17 polarization [71]. A ketogenic diet alters the gut microbiome, contributing to a decrease in intestinal Th17 cells [72] and alleviates DSS-induced colitis by inhibiting colonic ILC3s [73]. Eggerthella lenta, the human gut bacteria produce the Cgr2 enzyme, which induces IL-17a production in intestinal Th17 cells [74]. E. lenta are enriched in patients with IBD and exacerbate colitis in an RORγ-dependent manner in mice [74]. E. lenta-induced colitis can be inhibited by increasing dietary arginine levels, which in turn inhibits the effect of Cgr2 [74].
TREATMENT OF INFLAMMATORY BOWEL DISEASE BY REGULATION OF TH17 CELLS
Given the important role of Th17 cells in intestinal inflammation, neutralizing antibodies, and small molecules targeting Th17 cells and their associated cytokines may have therapeutic effects in controlling IBD (Table 1). Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, is approved by the FDA for the treatment of IBD [75]. In addition, selective IL-23-p19 inhibiting antibodies (risankizumab, brazikumab, guselkumab, and mirikizumab) have shown clinical benefits, including endoscopic remission and mucosal healing and are currently undergoing clinical trials [76–79]. IL-17A neutralizing mAbs (Secukinumab and ixekizumab) and IL-17A receptor-neutralizing mAbs (brodalumab) have exhibited opposite effects, with aggravation of IBD leading to premature termination of various trials [80–82]. Secukinumab, ixekizumab, and brodalumab are widely used for the treatment of multiple sclerosis, rheumatoid arthritis, psoriasis, and active ankylosing spondylitis [83]. The mechanisms underlying these paradoxical gastrointestinal effects are not well understood however, might suggest impaired intestinal epithelial barrier integrity, as IL-17A regulates epithelial tight junction proteins such as occludin. Vidofludimus is a novel oral immunomodulator that downregulates IL-17A, IL-17F, and IFN-γ levels by interference with the JAK/STAT3 and NFκB pathways [84]. Vidofludimus improved both acute and chronic DSS-induced colitis in mice [85] and patients with IBD [84]. These results suggest that inhibition of multiple cytokines may be effective in the treatment of IBD.
| Target | Drug | Experimental model or clinical trial status | Effective | Ref |
|---|---|---|---|---|
| IL-12/23 p40 | Ustekinumab | Phase III | Yes | [75] |
| IL-23p19 | Risankizumab | Phase III/II | Yes | [76] |
| Brazikumab | Phase III/II | Yes | [77] | |
| Guselkumab | Phase III/II | Yes | [78] | |
| Mirikizumab | Phase III/II | Yes | [79] | |
| IL-23 receptor | PTG-200 | Phase II/I | Yes | [86] |
| IL-17A | Secukinumab | Phase II | No | [80] |
| Ixekizumab | Phase III | No | [81] | |
| IL-17 receptor | Brodalumab | Phase II | No | [82] |
| IL-17A/IL-17F/IFN-γ | Vidofludimus | Phase I/II | Yes | [84] |
| IL-6 trans-signaling | Olamine (sgp130Fc) | Phase II | Yes | [87] |
| IL-6 receptor | Tocilizumab | Phase II | Yes | [88] |
| IL-1β | Canakinumab | Phase I (very early onset IBD) |
Yes | [89] |
| IL-1β receptor | Anakinra | Phase II | Yes | [90] |
| JAK inhibitor | Tofacitinib | FDA approved | Yes | [91] |
| α4β7 | Vedolizumab | FDA approved | Yes | [92] |
| TNF-α | Infliximab, Adalimumab, Certolizumab | FDA approved | Yes | [93] |
Gut microbiota and its metabolites play a role in maintaining gut barrier integrity and IBD by regulating Th17/Treg cell development and activity. Thus, the interactions between the gut microbiota and Th17/Treg cells may be promising targets for the treatment of IBD. Studies have shown that FMT, probiotics, and plant extracts are a potential therapeutic solution for IBD by targeting the gut microbiota based on the Th17/Treg cell balance (Table 2).
| Type | Drug or method | Effect | Ref | |
|---|---|---|---|---|
| Th17/Treg ratio | Microbiota | |||
| Plant extract | Rabdosia serra | Reduce | Lactobacillus ↑ Bacteroidetes ↑ | [5] |
| Stigmasterol | Reduce | Clostridia, Helicobacter ↑ Streptococcus ↓ | [94] | |
| Juglone | Reduce | Firmicutes ↑ Bacteroidetes ↓ | [95] | |
| Parthenolide | Reduce | Alloprevotella ↑ Bacteroides ↓ | [96] | |
| Probiotics | Lactobacillus paracasei R3 | Reduce | - | [97] |
| Lactobacillus rhamnosus GG | Reduce | - | [98] | |
| Lactobacillus acidophilus | Reduce | - | [99] | |
| Carbohydrate | Pectic oligosaccharides | Reduce | Bacteroidetes ↑ Firmicutes ↓ | [100] |
| Isomaltulose | Reduce | Bacteroidetes ↑ Firmicutes ↓ | [101] | |
| Fatty acid | Linoleic acid | Reduce | Bacteroidetes ↑ Firmicutes ↓ | [6] |
| Butyrate | Reduce | - | [102] | |
CONCLUSION AND PERSPECTIVE
Intestinal Th17 cells and the cytokines they produce, play critical roles in the pathogenesis of IBD. These results are being actively studied for the development of immunotherapies for IBD. In addition, gut microbiota-based therapies are consistently being developed for the prevention and treatment of IBD. However, several questions remain unanswered. More fundamental and translational studies are required to further understand the relationship between gut microbiota and mucosal immunity. Adjusting the gut microbiome, selecting certain bacteria, and performing FMT in conjunction with biological therapies may improve the diagnosis, and aid in prevention, and treatment of IBD.