





Introduction
Prednisolone is widely used as anti-inflammatory and immunosuppressive agent for variety diseases, and they are associated with many side effects such as hyperglycemia in humans [1]. Glucocorticoids (GCs) administration has been reported to be involved in the development of transient or permanent diabetes mellitus (DM) in dogs that presumably have pre-existing beta-cell defect, islet pathology, or both [2–4], but there is a lack of information on pancreatic histopathology in dogs with GC-induced DM.
Diabetic ketoacidosis (DKA) is thought to result from a relative or absolute decrease in endogenous serum insulin concentration along with an increase in insulin counter-regulatory hormones due to concurrent diseases [5]. This hormonal imbalance may contribute to increased peripheral lipolysis and ultimately to the production of the ketone bodies consisting of acetoacetate, beta-hydroxybutyrate, and acetone. Because GCs affect the hormonal imbalance, peripheral lipolysis, and the ketone body production, it could solely induce hyperglycemia as well as DKA. Indeed, 13 of 127 dogs with DKA were receiving oral or injectable GCs at the time or within 2 weeks of evaluation for DKA in a previous study [6], but the information of dose, type, and indication of the GCs was not discussed due to the nature of the study. Therefore, there is still lack of information that the prednisolone-induced DKA in dogs. Furthermore, there is no data for the permanent hyperglycemia without pancreatic pathology after resolution of DKA in dogs treated with long-term prednisolone administration. The purpose of this case report is to describe the clinical course of DKA in a dog with long-term therapeutic prednisolone administration for meningoencephalitis of unknown etiology (MUE), and to present the histopathologic findings of the pancreas in this dog with sustained insulin therapy two years following DKA resolution.
Case Report
A one-year-old intact male Maltese weighing 3.30 kg presented with status epilepticus. The dog was up to date on routine vaccinations. The dog had a history of salivation and right palpebral twitching. Brief neurologic examination revealed an absent menace response in the left eye and a right side head turn. Magnetic resonance imaging was performed in order to examine the etiology of seizure. Poorly-marginated and diffuse T2 weighted and fluid- attenuated inversion recovery hyperintensity of the grey matter was observed in the right occipital lobe of the cerebrum, adjacent cerebellum, and cerebellar flocculus. These areas were not enhanced after IV contrast administration. Cerebrospinal fluid analysis revealed lymphocytic pleocytosis. Based on the above results, MUE was tentatively diagnosed. This was managed with prednisolone (Solondo tab., Yuhan, Seoul, Korea; 0.6–1.0 mg/ kg PO 12 hourly), phenobarbital (Phenotal tab.; Daehan New Pharm, Hwasung, Korea; 2.5 mg/kg PO 12 hourly), and intermittent cytosine arabinoside (Cytosar-U inj.; Pfizer, New York, NY, USA; 50 mg/m2 IV 12 hourly), along with monitoring of clinical signs.
After three months, the dog was referred with dehydration, anorexia, and marked hyperglycemia. The dog was lethargic but responsive. The dog weighed 3.04 kg and had a body condition score of 3/9. Rectal temperature was 37.9°C, heart rate was 132 beats/min, and respiratory rate was 36 breaths/min. Moderate loss of skin turgor, dry oral mucous membrane, and slight prolongation of capillary refill time (> 2 seconds) were detected upon physical examination, which was consistent with moderate (up to 8%) dehydration. The blood glucose and 3-beta-hydroxybutyrate concentrations were 504 mg/dL [reference interval (RI): 65 to 118 mg/dL] and 4.1 mmol/L [> 3.8 mmol/L for diagnosis of DKA [7]], respectively. Venous gas analyses revealed a pH of 7.296 (RI: 7.350 to 7.450), HCO3– of 16.7 mmol/L (RI: 15 to 23 mmol/L), PCO2 of 34.2 mmHg (RI: 35.0 to 38.0), and base excess of –10 mEq/L (RI: –5 to 5 mEq/L), which were consistent with metabolic acidosis with compensatory respiration. Urine pH was 6.0, specific gravity was 1.016, urine protein (30 mg/dL), with glucose of 500 mg/dL, ketone of 6.0 mmol/L, 3 to 5 red blood cells (RBC)/hpf (RI: 0 to 5 /hpf), and 1 to 3 white blood cells (WBC)/hpf (RI: 0 to 5 /hpf). Based on all of this, DKA was initially diagnosed, and further examinations, including a complete blood count, serum biochemical profile, SNAP cPL test (IDEXX Reference Laboratory, Seongnam, Korea), serum electrolytes analyses, radiography, and abdominal ultrasonography, were performed.
The abnormalities of complete blood counts, serum biochemistry, and electrolytes were presented in Table 1. There was a moderate regenerative left shift (1.4 × 109/L; RI: < 0.3 × 109) with a few neutrophils exhibiting mild toxicity (cytoplasmic basophilia and/or Döhle bodies) in blood film with haematoxylin and eosin stain. The result of the SNAP cPL test was negative. Endocrine testing revealed a serum insulin concentration of 14.0 μIU/mL (RI: 5.2 to 41.5 μIU/mL). In addition, insulin sensitivity and the role of pancreatic β-cells were assessed by homeostatic model assessment (HOMA) indices [8]. HOMAinsulin sensitivity and HOMAβ-cell function were calculated using a non-linear formula on a dedicated calculator (HOMA Calculator Version 2.2.3, Diabetes Trial Unit, University of Oxford, Oxford, UK). HOMAinsulin sensitivity and HOMAβ-cell function showed a lower value than that of age-matched healthy dogs (Table 2). Abdominal and thoracic radiographs were within the normal limits. Abdominal sonography showed a hyperechoic and homogenous liver parenchyma. Other organs were within normal limits. Upon examination, no sources of infection or inflammation were identified.
* Blood glucose levels were determined using an automated analyzer (Hitachi; Hitachi High-Technologies, Tokyo, Japan).
** Insulin levels were determined using a chemiluminescent immunoassay-based autoanalyzer (Immulite 1000; DPC, Los Angeles, CA, USA).
† HOMA indices were calculated with nonlinear formulas in HOMA calculator (HOMA Calculator Version 2.2.3; Diabetes Trial Unit, University of Oxford, Oxford, UK) validated in dogs. In some cases in which glucose or insulin concentrations exceeded value allowed by the HOMA calculator, the mathematical formula was used to calculate the HOMA indices:
The dog was hospitalized and treated with 0.9% normal saline (0.9% NaCl; Daihan Pharm, Seoul, Korea) and a regular insulin (Humulin R; Eli Lilly and Company, Seoul, Korea) constant rate infusion that was prepared by adding 4.4 U/kg regular insulin to 500 mL of 0.9% normal saline and administered at a rate of 5 to 10 mL/hour. Cefotaxime sodium (Cefotaxime Hanmi Inj.; Hanmi Pharm, Seoul, Korea, 30 mg/kg IV 12 hourly) as prophylactic antibiotics was also administered. Insulin resistance induced by prednisolone could potentially antagonize the treatment of DKA, but this therapy could not be ceased due to the management of MUE.
On day 4, the blood glucose and 3-beta-hydroxybutyrate were almost normalized. The demeanor of the dog improved and voluntary feeding was observed. The next day, the dog was discharged with medications including 0.3 units/kg of recombinant human neutral protamine Hagedorn (NPH) insulin (Humulin N; Eli Lilly and Company, Seoul, Korea) and phenobarbital (2.5 mg/kg PO 12 hourly). Because of concern that the prednisolone might antagonize insulin therapy, the dose of prednisolone was reduced to 0.4 mg/kg PO 12 hourly. The blood glucose curve was stabilized 72 days after insulin therapy.
Over the next two years, the dog continued to be routinely re-evaluated and was managed with permanent insulin therapy (0.8–1.4 units/kg SC 12 hourly) and medications including prednisolone (0.4–1.1 mg/kg PO 12 hourly). The doses of insulin and prednisolone were periodically adjusted based on the results of history, physical and neurologic examination findings, serial blood glucose curves and blood analyses including serum fructosamine concentrations (321 to 586 μmol/L) and the activity of liver enzymes such as alanine transaminase (142 to 329 U/L; RI = 10 to 100) and alkaline phosphatase (267 to 3,193; RI = 23 to 212). The dog was euthanized after two years because of uncontrolled status epilepticus.
A pancreas sample was obtained following euthanization with the owner’s consent. Slides stained with hematoxylin and eosin were examined. The sections of pancreas that were examined were within normal limits (Fig. 1). There was no evidence of neoplasia, inflammation, or necrosis within the pancreas or the surrounding peripancreatic adipose tissue. There was also no evidence of vacuolization, fibrosis, or vascular or ductal changes in pancreatic islets.
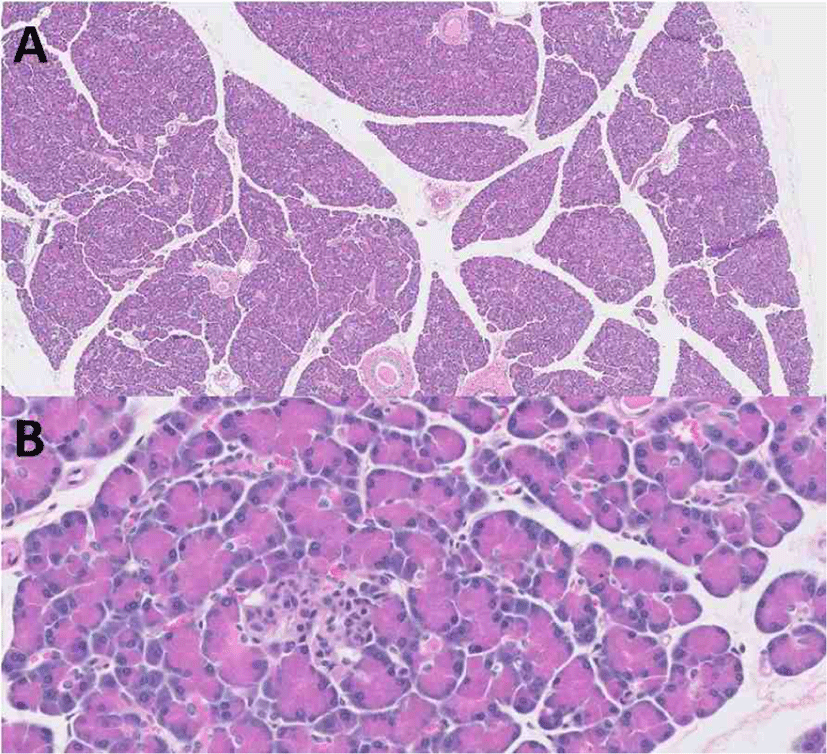
Discussion
This is a case report of a dog with MUE that developed DKA in association with prednisolone therapy. Upon presentation, HOMA indices were lower than that of age-matched dogs and post-mortem histopathologic examination of the pancreas revealed that the pancreas was within normal limits, indicating that insulin resistance induced by prednisolone therapy may have caused DKA and permanent hyperglycemia without histopathologic abnormalities of the pancreas in the present case.
DKA is a common life-threatening complication of DM in dogs [9]. The development of DKA is thought to result from an absolute or relative insulin deficiency with concurrent disease which results in increased insulin counter-regulatory hormones [10, 11] and/or dysregulation of pro-inflammatory cytokines [12]. It was possible that pre-existing MUE was involved in the pathogenesis of DKA in the present case as it may dysregulate the production of inflammatory cytokines [13], which can contribute to the impairment of glucose homeostasis and/or the increase of insulin counter-regulatory hormones. Dysregulated serum pro-inflammatory cytokines, such as tumor necrosis factor receptor-1, interleukin-1β, tumor necrosis factor-α, and interleukin-6, were identified before and after treatment in human type 2 DM patients with hyperglycemic crisis, implying that an inflammatory process can cause further impairment of glucose metabolism in insulin resistance status [14], and dysregulation of pro-inflammatory cytokines were also identified in dogs with DKA [12]. In the present case, there may be an inflammatory process supported by a moderate regenerative left shift with a few neutrophils exhibiting mild toxicity. Furthermore, the clinical signs (such as seizure) were progressive and the doses of prednisolone had been gradually increased. Therefore, it was possible that the progression of MUE dysregulated the pro-inflammatory cytokines and increased insulin counter-regulatory hormones, which subsequently induced the onset of DKA in combination with prednisolone-induced insulin resistance (relative insulin deficiency), lipolysis, and ketogenesis. In addition, the dysregulation of pro-inflammatory cytokines and insulin counter-regulatory hormones due to the progression of MUE may have contributed to the life-long need for insulin therapy in this case.
The dog reported here had been diagnosed with MUE and had been managed with medications including prednisolone for three months. The term MUE refers to a probable meningoencephalomyelitis in which an infectious cause has not been identified yet, and that lacks a histopathological diagnosis [15]. This term is widely used because definitive ante-mortem diagnosis of the underlying disorder is unlikely without biopsy, which can be difficult in practice. Although MUEs are classified as granulomatous meningoencephalomyelitis, necrotizing meningoencephalomyelitis, and necrotizing leukoencephalitis, they may lead to aberrant immune-mediated response against the constituents of the central nervous system [16]. Aggressive immune-suppression has been considered the mainstay therapy for MUE. Specific therapeutic protocols have not yet been established [16]. There are inconsistent results regarding the benefit of combined treatment using corticosteroids and other immunosuppressive drugs compared to prednisone alone [16, 17], but it has been generally accepted that the addition of other immunosuppressants such as mycophenolate mofetil, azathioprine, and cyclosporine to a corticosteroid regimen is beneficial because this permits for a reduction of the prednisone dose, and subsequently, of its adverse effects [15‒17]. In the present case, the addition of cyclosporine was initially considered, but cyclosporine has been shown to suppress insulin secretion during in vivo assessments of canine pancreatic islet cells [18] which has also been identified in healthy dogs [19], therefore this agent was not used. Alternatively, azathioprine was also considered. In a previous study, azathioprine appeared to be a safe and potentially effective adjunct to corticosteroid for the treatment of dogs with MUE [17]. However, the occurrence of DM was also identified although it was a less common adverse effect and may have been attributable to the concurrent corticosteroid administration [17]. Therefore, mycophenolate mofetil (Cellcept cap.; Roche Laboratories, Nutley, NJ, USA, 20–25 mg/kg PO 12 hourly) was added as an adjunctive treatment with prednisolone for MUE in the present case. Anticonvulsive medications such as zonisamide (Excegran tab.; Eisai Korea, Seoul, Korea, 25 mg/kg PO 12 hourly) and phenobarbital (2–4 mg/kg PO 12 hourly) were also prescribed to manage seizure.
The dog in the present report received a long-term therapeutic dose (0.6–1.0 mg/kg PO 12 hourly) of prednisolone, which can unintentionally impair glucose homeostasis. Prednisolone is a synthetic GC that has been used in veterinary practice because of its anti-inflammatory and immunosuppressive functions. However, GCs can affect pancreatic beta-cell function by decreasing insulin secretion [20, 21] and can also induce insulin resistance by directly interfering with several steps in the insulin signaling pathway, resulting in reduced glucose uptake and glycogen synthesis in the rat skeletal muscle and liver [22]. In order to enhance insulin sensitivity, the pancreatic beta-cell generally increases insulin secretion to maintain normal glucose levels, but HOMAβ-cell function was low in the present case, although hyperglycemia occurred after prednisolone administration. Prednisolone has some different characteristics comparing to dexamethasone which has been widely studied as diabetogenic drug, but HOMA indices revealed impaired pancreatic beta-cell function and reduced insulin sensitivity in the present case treated with prednisolone which can induce hyperglycemia after long-term administration in humans [1]. In the present case, HOMA indices were used for assessing insulin sensitivity, but it generally measures inhibition of glucose output (hepatic insulin sensitivity) more than glucose uptake (peripheral insulin sensitivity) in fasting status [8], thus it can be a limitation that dynamic tests such as glucose tolerance tests or euglycemic hyperinsulinemic clamps were not performed, but these procedures can be unusual in clinical practice [23].
It has also been previously reported that chronic GC administration could induce rat pancreatic beta-cell apoptosis [24]. GC-induced apoptosis in insulin-secreting cells is accompanied by a downregulation of Bcl-2, activation of calcineurin with subsequent dephosphorylation of the proapoptotic protein of the Bcl-2 family, and mitochondrial depolarization, which may reduce beta-cell mass in the long term [24]. It is believed that the administration of GCs may cause overt DM in dogs that presumably have pre-existing pancreatic islet pathology [4]. A previous study revealed a massive reduction of total islet endocrine cells and beta-cells in cases of both new and longstanding canine DM [25]. This was obvious in histopathology of hematoxylin- and eosin-stained slides. However, histopathologic examination of the pancreas was within normal limits in the dog reported herein, indicating that pancreatic beta-cell dysfunction occurred without apoptosis or a reduction of beta-cell mass, and that sustained insulin resistance could result in a prolonged need for exogenous insulin therapy along with prednisolone administration after resolution of DKA. This assumption would be supported by discontinuing prednisolone and looking for a reversal of the insulin resistance or hyperglycemia, but this could not be performed in the present case because the dog needed to be managed with prednisolone therapy for MUE, which could induce a life- threatening condition if left untreated. Further study is needed to understand the histopathology in prednisolone-induced hyperglycemia or DKA through more advanced methods such as the morphometry of individual islets with high-resolution high-magnification fluorescent microscopy and antisera against islet hormones [25].
Glucotoxicity refers to structural and functional damage in pancreatic beta-cells and the target tissues of insulin caused by chronic hyperglycemia. Dogs can be considered as sensitive to glucose toxicity and lose their beta-cell mass quickly if challenged with hyperglycemia [26]. It can be reversible in the early stages with aggressive treatment to normalize blood glucose concentration [27]. In the present case, hyperglycemia was relatively well controlled 72 days after initial presentation, indicating that the duration of the dog exposure to glucotoxicity might be a minimum of 72 days. Therefore, it is possible that the time was not sufficient to cause permanent beta-cell pathology.
In conclusion, GC-induced DM has been recognized as a complication of GC use for over 60 years and is considered a harmful problem, but scientific evidence and strategies for prevention and treatment are lacking in human and veterinary medicine [28]. In addition, acute moderate to high dose of prednisolone can cause fasting hyperglycemia, but chronic low-dose of prednisolone can not increase in blood glucose level in human patients without known DM [1]. However, the effects of GCs on blood glucose levels are dose dependent, it might be possible that chronic moderate to high dose of prednisolone cause fasting hyperglycemia. Permanent DM after GCs administration have been also noted sporadically in dogs [3, 29], and severe complications including DKA can occur as described herein. Rapid recognition of the occurrence of hyperglycemia after long-term prednisolone administration and proper strategies for reducing glucose levels such as insulin sensitizer and exogenous insulin can prevent DKA and restore pancreatic pathology, especially if the dog has an inflammatory disease that is progressive and needs life-long prednisolone therapy. This case report presents not only the clinical feature, but also important bases for future studies investigating the pathophysiology of prednisolone-induced DM in dogs.