





Introduction
The historical backdrop of herbal medication is as old as human civilization [1]. Medicinal plants have been utilized as a constant source of alleviate a variety of diseases [2] and antiviral effects of medicinal plants have been investigated as remedies for different ailments [3]; however, while most of the herbal remedies are appreciated within the local communities, most of them lack documented evidence of prophylactic and therapeutic effectiveness [4]. Therefore, many medicinal plants are now being collected and examined to identify possible sources of antiviral agents [5–7].
Several types of viruses cause destructive diseases and outbreaks worldwide, resulting in significant mortality and economic damage. For instance, influenza A viruses have been accountable for seasonal epidemics and have led to three pandemics in the last few decades (1918, 1957, and 1968) [8]. Another example is SARS-CoV, which is an acute and non-resolved disease that can be harmful with a 3% case fatality rate [9]. Although lots of contraceptive or therapeutic drugs and vaccines have been developed to control, prevent, and treat viral diseases, the emergence of novel mutants or resistant virus strains diminishes their efficacy and causes public health problems. Therefore, different studies have attempted to identify novel components with antiviral functions from natural or synthetic resources [10]. As antiviral substances, natural products are major valuable sources as many studies have been able to identify antiviral substances from natural sources with high efficacy, less toxicity, and minor side effects [11].
Fossil evidence has acknowledged that human use of flora as folk medicines back at least 60,000 years [12]. According to the World Health Organization (WHO), almost 65% of the world’s population stated make use of natural ingredients as medicinal components [13]. Roughly around 20,000 plant species utilized for medicinal purposes have been accounted by WHO [14]. The use of present-day analytical technologies applied towards the active compounds found in medicinal plants have allowed for greater insights into plant-derived pharmaceutical materials [15].
The tree Melia azedarach (Family: Meliaceae) is locally recognized as bakain or drek (Hindi), Persian lilac, or China tree (English), and Fleurs lilas (French). In South America, it is generally known as “paraiso” or paradise, and in the US as Indian lilac or white cedar [16]. The whole plant or its specific parts (leaves, stem, and roots) are known to have medicinal properties and have an extended history of use by indigenous and tribal people in India. The extract is used as an ayurvedic medicine in India and Unani medicine in Arab countries as its antioxidative, analgesic, anti-inflammatory, insecticidal, rodenticidal, antidiarrheal, deobstruent, diuretic, antidiabetic, cathartic, emetic, antirheumatic, and antihypertensive activities [17]. Although there are numerous reports on the immunomodulatory properties of Melia azedarach, the broad spectrum of its antiviral effects has not been fully explored [18].
This study aimed to clarify the preventive and therapeutic effects of Melia azedarach extract (MAE) on a broad spectrum of viruses in cell lines. Furthermore, we identified that the herb extract treatment could induce pro-inflammatory cytokines, interferons, and interferon-stimulated gene transcription level in immune and epithelial cell lines. Finally, we confirmed the prophylactic and therapeutic effects of MAE in vivo.
Materials and Methods
Crude plant material of Melia azedarach was purchased from a local store (Jaecheon Oriental Herbal Market, Jaecheon, Korea) and verified by Professor Ki-Hwan Bae at the College of Pharmacy, Chungnam National University. The water soluble herbal extract of Melia azedarach, was prepared by Vitabio Corporation, Daejeon, Korea. First, Melia azedarach (100 g) immersed in 1 L of distilled water and extracted by heating for 2.5 hours at 105°C. Then the extract was filtered using a filter paper (0.45 μm) and stored at 4°C for 24 hours. After the extraction proses, the fluid concentrate of herbal extract was centrifuged at 14,240×g for 15 min and the pH of the collected supernatant was adjusted to 7.0. Finally, the total aqueous extract was filtered by using membrane syringe (0.22 μm) and successive aqueous extract was stored at −20°C until further use.
RAW264.7 cells (ATCC TIB-71), HEK293T (ATCC-11268) and HeLa (ATCC CCL-2) cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, Carlsbad, CA, USA) enriched with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and 1% antibiotic/antimycotic solution (Gibco, Grand Island) with 37°C under 5% CO2 in a humid incubator.
Green Fluorescence Protein (GFP) fused Influenza A/PuertoRico/8/34(H1N1) (PR8-GFP) and challenge viruses [{A/Aquaticbird/Korea/W81/2005(H5N2)}, {A/PR/8/34 (H1N1)}, {A/Aquaticbird/Korea/W44/2005(H7N3)} and {A/Chicken/Korea/116/2004(H9N2)}] were provided by Dr. Y. K. Choi, Chunbuk National University, Cheongju, Korea and were amplified in the allantoic fluid of 10-day-old embryonated chicken eggs. Herpes simplex virus (HSV-GFP) was kindly gifted from Dr. Jae U. Jung, Department of Molecular Microbiology and Immunology, University of Southern California, USA and propagated in confluent Vero cells (ATCC CCL-81). Coxsackie virus (H3-GFP) and EV71 were propagated in confluent HeLa cells while bovine rhinovirus (BRV) was amplified in confluent ZZR cells. The resulting viral suspension was aliquot, stored at −70°C and titer was determined by a standard plaque assay and TCID50.
A virus replication inhibition assay was performed as previously describe in [19], with some modifications. Briefly, RAW264.7 (1 × 106 cells/well), HEK293T and HeLa cells (8 × 105 cells/well) were cultured in 6-well plates and were incubated at 37°C for 12 hours. Cells kept DMEM alone (untreated and virus-only groups), or treated with 1,000 U recombinant mouse or human IFN-β (Sigma-Aldrich, St. Louis, MO, USA) or with 10 ng/mL MAE. After 12 hpt, cells were infected with either PR8-GFP, HSV-GFP, or H3-GFP using DMEM containing 1% FBS. At 2 hpi, wells were washed with autoclaved phosphate buffered saline (PBS) and replaced the media with DMEM (10% FBS). Cells and cell supernatants were collected for virus titer determination at 12 and 24 hpi.
HeLa cells, HEK293T and Vero (8 × 105 cells/well) cells were cultured in 6-well tissue culture plates and incubated at 37°C for 12 hours. DMEM alone (untreated and virus-only groups), 1,000 U recombinant mouse or human IFN-β, or 10 ng/mL MAE were co-treated with either PR8-GFP, H3-GFP or EV71 infection. At 2 hpi, replaced the media with DMEM (10% FBS). Samples were collected for virus titer determination at 12 and 24 hours post-infection (hpi).
HEK293T and HeLa cells were cultured in 6-well plates (8 × 105 cells/well) and incubated at 37°C for 12 hours. Cells were infected with either H3-GFP, BRV, or EV71 using DMEM containing 1% FBS. At 2 hpi, wells were washed with autoclaved PBS and replaced the media with DMEM (10% FBS). After 2, 4, or 6 hpi cells were left with DMEM alone, or treated with 1,000 U recombinant mouse and human interferon IFN-β or 10 ng/mL MAE and incubated 12 hour. Samples were collected for virus titer determination at 12 hpi.
Virus titers were determined by standard plaque assay as described before using Vero cells with some modifications [20]. Briefly, Monolayers of Vero cells (5 × 105 cells/well) were seeded in 12-well tissue culture plates. After 12 hours incubation, cells were inoculated with serially diluted viral suspensions for 2 hours. Following 2 hours of incubation at 37°C, the inoculums were replaced with 10% FBS containing DMEM with agar (0.45 g/20 mL). Then plates were incubated for another 46 hours at 37°C and examined for plaque formation. Viral titers ware calculated using the number of plaque-forming units (PFU) and the dilution factor. In the case of PR8-GFP titration, both cell supernatant and infected cells from each groups were collected at 12 and 24 hpi separately and subjected to alternative freezing and thawing under room temperature and −70°C for 5 minutes for five repeated cycles. Then, those cells were re-suspended with 200 µL of PBS and dilution series were performed before infecting Vero cells.
Viral titers were measured by median tissue culture infectious doses (TCID50) using Vero and HeLa cells for EV71 and BRV respectively [21]. Confluent Vero and HeLa cells grown in 96-well microtiter plates were infected with 10-fold serial dilutions (in DMEM containing 1% FBS) of harvested supernatants (50 μL/well) of EV71 and BRV, respectively. After 2 hour at 37°C in a humid atmosphere with 5% CO2, the inoculums ware replaced with DMEM containing 10% FBS and incubated for another 48 hours. Cytopathic effect of the viruses was observed daily and titers were determined by CPE-TCID50.
The pro-inflammatory cytokines-induced by MAE on RAW264.7 cells and HEK293T cells were examined using commercial ELISA kits for murine and human interleukin (IL)-6 (BD Bioscience, La Jolla, CA, USA) and IFN-β (PBL Interferon Source, Piscataway, NJ, USA) as manufacturer protocol. Briefly, RAW264.7 cells (1 × 106 cells/well) and HEK293T (8 × 105 cells/well) were cultured in 6-well tissue culture plates. After 12 hours, cells were treated with 1,000 U recombinant mouse or human interferon (IFN)-β, 10 ng/mL MAE in DMEM containing 10% FBS, then incubated at 37°C with 5% CO2. Supernatants were collected at 12 and 24 hpt, and clarified by centrifugation at 598×g for 10 min at 4°C and dispensed into murine or human IFN-β ELISA plate or IL-6 capture antibody-coated ELISA plates. Murine IFN-β ELISA was performed in duplicate and other IL-6 was performed in triplicate.
RAW264.7 cells (1 × 106 cells/well) and HEK293T cells (8 × 105 cells/well) were seeded in 6-well tissue culture plates and incubated at 37°C. After 12 hours, cells were left untreated (negative control) or treated with 1,000 U recombinant mouse or human IFN-β (positive control) or 10 ng/mL MAE and cells were harvested at 0, 8, and 12 hpt. Total mRNA was extracted and Complementary DNA (cDNA) synthesis was accomplished using reverse-transcriptase PCR (Toyobo) [22]. The cDNAs were quantified by SYBR (Qiagen) based RT-PCR using various cytokine-specific primers and normalized to GAPDH and β-actin in RAW264.7 and HEK293T cells respectively. The transcription level of mRNA was obtained by the 2−ΔΔCt method as described previously [23]. Results were expressed as fold induction. The RT-PCR primers are listed in Table 1.
Phosphorylation of type I IFN and NF-κB related proteins were determined by immunoblot analysis [24]. Briefly, RAW264.7 cells were seeded on 6-well tissue culture plates (1 × 106 cells/well) and incubated at 37°C. After 12 hours, cells were left untreated (negative control), or treated with 100 ng/mL LPS (Invitrogen™ 00-4976-93) (positive control) or 10 ng/mL MAE and cells were harvested at 0, 8, 12, and 16 hpt. The cell pellets were washed once with ice cold PBS. Next, cell pellets were lysed in Radio-immunoprecipitation assay (RIPA) lysis buffer (50 mM Tris-HCl [pH 8.0]), 150 mM NaCl, 0.5% sodium deoxycholate, 1% IGEPAL, 1 mM NaF, 1 mM Na3VO4, and 1 μg/mL each of aprotinin, leupeptin). Whole-cell lysates (WCL) were mixed with a 10x sample buffer (S3401-1VL, Sigma-Aldrich) at 1:1 ratio to separate by SDS-PAGE and transferred onto a PVDF membrane (IPVH00010, Bio-rad) in a buffer containing 30 mM Tris, 200 mM glycine, and 20% methanol for 2 hours. Membranes were blocked for 1 hour in Tris-buffered saline containing 0.05% Tween 20 and 5% Bovine serum albumin (TMS013,Sigma-Aldrich) and then probed with target protein antibody in 5% FBS–TBST at 4°C overnight with anti-IRF3 (#4302), anti-phospho-IRF3 (Ser396, #4947), anti-STAT1 (#9172), anti-phospho-STAT1 (Tyr701, #7649), anti-p65 (#4764), anti-phospho-p65 (Ser 536, #3033), anti-TBK-1 (#3504S), anti-phospho-TBK-1 (Ser172, #5483), anti p44/42 (#9102S), anti-phospho p44/ 42 (Thr202/Tyr204, #9101), anti-p38 (#9212), anti-phospho-P38 (Thr180/Tyr182, #9211) (Cell signaling,) and β-actin (sc-47778, santa cruz). After three (10-min) washes with Tris-buffered saline containing 0.05% Tween 20, the membranes were reacted with a horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. After three (10-min) washes with TBST, the reaction of HRP was visualized with an enhanced chemiluminescence detection system (EZ-western Lumi Femto, DOGEN) using a Las-3000 mini lumino-image analyzer.
Five-week-old 80 female BALB/c mice purchased from orient bio (Korea) and separated into 4 experimental sets (four Virus strains), with 4 groups each containing 5 mice per one group. Mice were orally administered 0.1 mg/mL MAE in a total volume of 100 µL (10 µg per head) at 1, 3, and 5 days before infection and 1, 3, 5, and 7 days after infection. Mice in control groups were inoculated with same dose of autoclaved PBS. Then, Mice were anesthetized with ketamine for a short time period, and intranasally challenged with 5 times of 50% mouse lethal dose (5MLD50) of H1N1, H5N2, H7N3, or H9N2 in 20 μL PBS per mouse. Treatment and challenge experiments were performed in an approved BSL-2+ facility. Body weight variation and survival rates were observed up to 13 dpi. Mice exhibiting more than 25% of body weight loss were considered to have reached the experimental endpoint and were humanely euthanized.
Five-week-old 48 female BALB/c mice were separated into 2 experimental sets, with 4 groups per each set containing 6 mice per group. Mice were orally administered 0.1 mg/mL MAE in a total volume of 100 µL (10 µg per head) at 1, 3, and 5 days before infection (pre-treatment) and 1, 3, 5, and 7 days after infection (post-treatment) and both treatment together (Pre and post treatment). Mice in control groups were inoculated with same dose of autoclaved PBS. Mice were anesthetized with ketamine for a short time period, and intranasally challenged with 3 times of 50% mouse lethal dose (3MLD50) of H1N1 in 20 μL PBS per mouse. Treatment and challenge experiments were conducted in an approved BSL-2+ facility. Lung tissues from euthanized mice were collected aseptically at 3- and 5-days from the last inoculation (dpi). Lung viral titers were determined by median tissue culture infectious doses (TCID50) using Madin-Darby canine kidney (MDCK) cells as describe before [13].
Briefly, mice were sacrificed and the extracted lungs were homogenized in 500 μL PBS containing antibiotic/antimycotic compounds. Confluent MDCK cells cultured in 96-well microtiter plates were infected with 10-fold serial dilutions (in DMEM containing 1% FBS) of lung homogenate (50 μL/well). After 1hour at 37°C in a moist atmosphere with 5% CO2, the medium comprising L-1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK) trypsin (Thermo Fisher Scientific, Rockford, IL, USA) was added to the infected media and incubated for another 48 hours. Viral cytopathic effects were observed daily and Hemagglutination assay (HA) was performed to determine viral titer [25] as follows, Fifty microliters (50 μL) of 0.5% chicken red blood cells (RBC) was added to 50 μL of cell culture supernatant and incubated at room temperature for 30 min. Wells containing HA were scored as positive. The virus titer was calculated by the Reed and Muench method and expressed as Log10 TCID50/mL of lung tissues [26].
Statistical analysis was performed using Prism 6 (GraphPad Software). Data are expressed as the mean ± S.D. of at least two independent experiments. Statistical significance was analyzed using unpaired Student’s t-test as indicated in the legends. p<0.05, p<0.01 was regarded as significant.
Results
The antiviral effects of MAE were evaluated by observing the inhibition of viral replication in vitro. First, we determined the minimum concentration of MAE that shows the potent antiviral effect and non-cytotoxic effect to the treated cells from our preliminary study. Based on the results we selected 10 ng/mL dose of MAE treatment for our further in vitro experiments. Afterward, we determined viral replication in epithelial and macrophage cells pre-treated with MAE and subsequently infected with GFP-expressing viruses (Fig. 1A). HeLa and HEK293T cells were pre-treated for 12 h and infected with H3-GFP and HSV-GFP, respectively, followed by the determination of viral titers. The results showed a significant reduction in viral titer in the extract-treated group compared with that in the untreated group. A similar experiment was performed in the murine macrophage cell line. RAW264.7 cells displayed conspicuously reduced viral titers, whereas the untreated group had high viral titers in terms of PR8-GFP. Based on our findings, MAE showed excellent antiviral effect against H3-GFP, PR8-GFP and HSV-GFP (Fig. 1A). Moreover, these results suggest that pre-treatment with the herbal extract could inhibit viral replication in both epithelial and macrophage cell lines against DNA and RNA viruses.
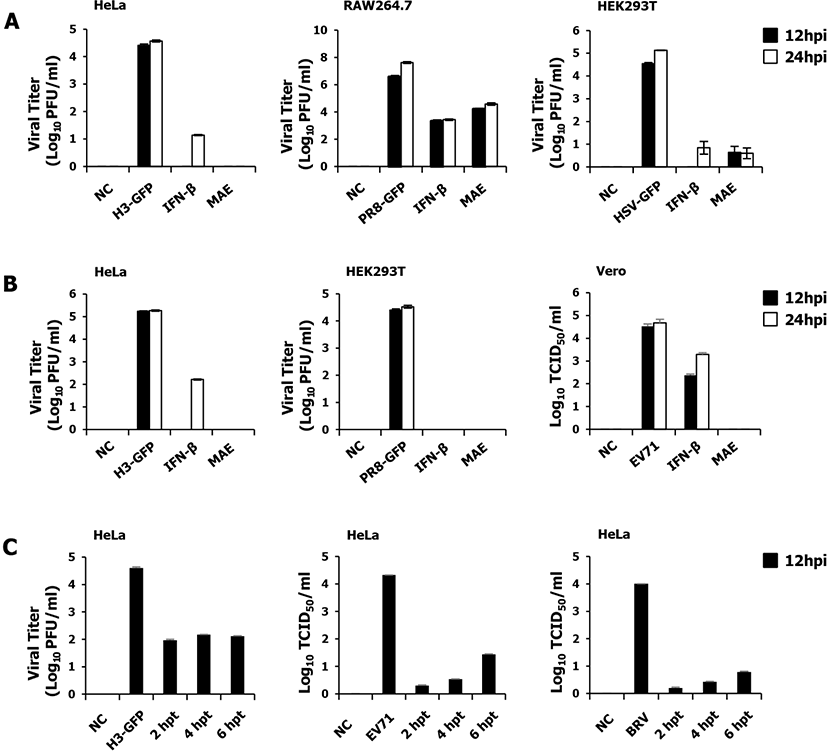
To evaluate the co-treatment antiviral effect of MAE, we simultaneously infected and treated the cells with the viruses and MAE. The anti-viral effect was determined with the H3-GFP on HeLa, PR8-GFP on HEK293T, and EV71 on Vero cells, respectively (Fig. 1B). As we have shown in the figure, treated cells exhibited a marked reduction in viral titer, whereas a high viral titer was observed in the untreated groups about all the viruses. The result of the co-treatment experiment was consistent with that of the pre-treatment result. These results distinctly show that the MAE can reduce the replication of the viruses in particular cell lines in co-treatment.
To determine the post-treatment antiviral effect of MAE, we treated the cells with MAE at 2, 4, and 6 hours following viral infection in HeLa cells (Fig. 1C). HeLa cells were infected with H3-GFP, EV71, and BRV. The results were indistinguishable to pre-treatment and co-treatment experiments. Viral replication was significantly reduced by MAE treatment at 2, 4, and 6 hpi compared with the non-treated group. The infected cells treated with MAE, rescued in a time-dependent manner. In the early stages, viral replication was strongly inhibited by MAE and increased slightly with time.
To determine whether MAE treatment could induce cytokine production and induce an antiviral immune response, the levels of the induced cytokines in RAW264.7 cells and HEK293T cells ware determined by ELISA. Moderate secretion of IL-6 and IFN-β were observed in RAW264.7 (Fig. 2A) and HEK293T (Fig. 2B) cells treated with MAE compared to the non-treated group. The antiviral effect of Melia azedarach could be related to the induction of innate immune response mediated by the expression of cytokines, such as IL-6 and IFN-β. To understand the link between these observations and the IFN-inducing signaling pathway, we examined the phosphorylation of interferon-related signal molecules and NF-κB signaling related molecules. To confirm the effects of MAE on the type I IFN signaling pathway, we performed immunoblot analysis using whole cell lysates of extract-treated RAW264.7 cells. As shown in Fig. 2C, MAE treatment significantly up-regulated the phosphorylation of IRF-3, P65, STAT1, TBK1, P38, and ERK which are key signaling molecules of type I interferon and NF-κB pathway. Once stimulation of Pattern Recognition Receptors (PRRs) of the host cell by LPS or Melia azedarach transduction of downstream signaling pathway is initiated, consequently interferon-stimulated genes (ISGs) are up regulated, which leads to induction of antiviral immune responses in the host cell. Our results demonstrate that treatment with MAE induced phosphorylation of type I IFN and NF-κB pathway molecules at 8 hpt, which dramatically increased with time. Furthermore, increased phosphorylation of STAT-1 indicates the active functions of ISGs. Besides the activation of type I interferons, extract-treated RAW264.7 cells could elicit an obvious activation of NF-κB (P65), leading to the strong secretion of pro-inflammatory cytokines. The secreted inflammatory cytokines are activated the immune cells and act on the rapid clearance of viruses [27]. Interestingly, the phosphorylation of these molecules by MAE is comparable to that by LPS treatment, which is a known potential stimulator of TLR4.
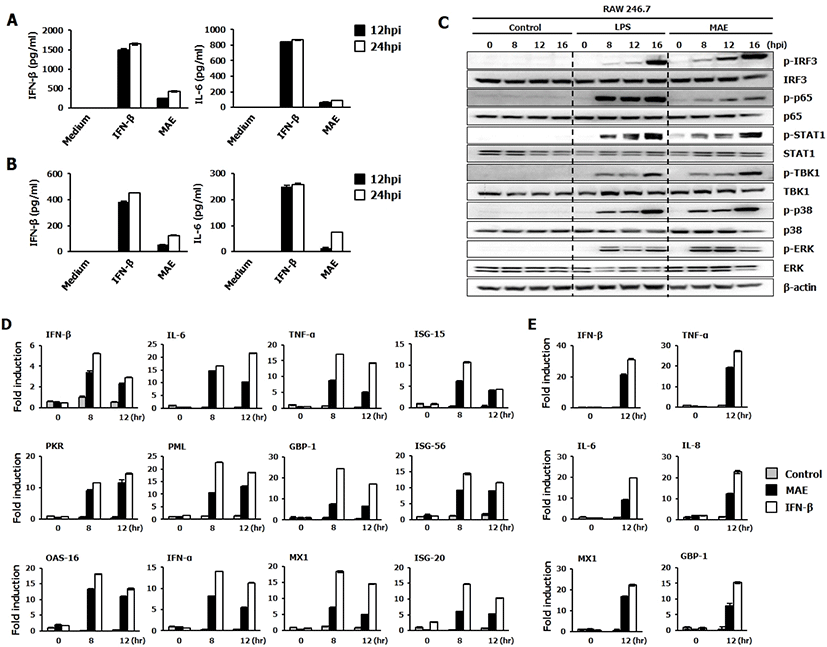
Based on the levels of secretion of cytokines and phosphorylation of signaling molecules, we further evaluated the induction of different antiviral related genes and ISGs at the transcriptional levels in response to MAE treatment in RAW264.7 and HEK293T cells by using quantitative polymerase chain reaction (Fig. 2D–E). As confirmed by the qPCR, the expression of ISG and antiviral genes were upregulated compared to the non-treated cells. Extract treated cells displayed a significant high mRNA gene transcription fold induction of IFN-β, IL-6, TNF-α, ISG-15, PKR, PML, GBP-1, ISG-56, OAS-16, IFN-α, MX1 and ISG-20 compared to the non-treated cells at 8 and 12 hpt in the extract-treated RAW264.7 cells (Fig. 2D).
Furthermore, comparable transcriptional activation patterns were observed in extract-treated HEK293T cells (Fig. 2E). Cells treated with MAE exhibited a similar pattern to IFN-β treated cells (positive control) after normalization to β-actin. Interestingly, HEK293T cells treated with MAE showed increased expression of human IFN-β, TNF-α, IL-6, IL-8, MX1 and GBP-1 genes at 12 hpt compared to the non-treated cells (Fig. 2E). The overall results demonstrate that MAE could up-regulate the transcription levels of IFN-β, IL-6, and various ISGs in both macrophages and epithelial cells. This molecular-level stimulation may have a direct correlation with the antiviral effects of the extract, which were detected in both RAW264.7 and HEK293T cells.
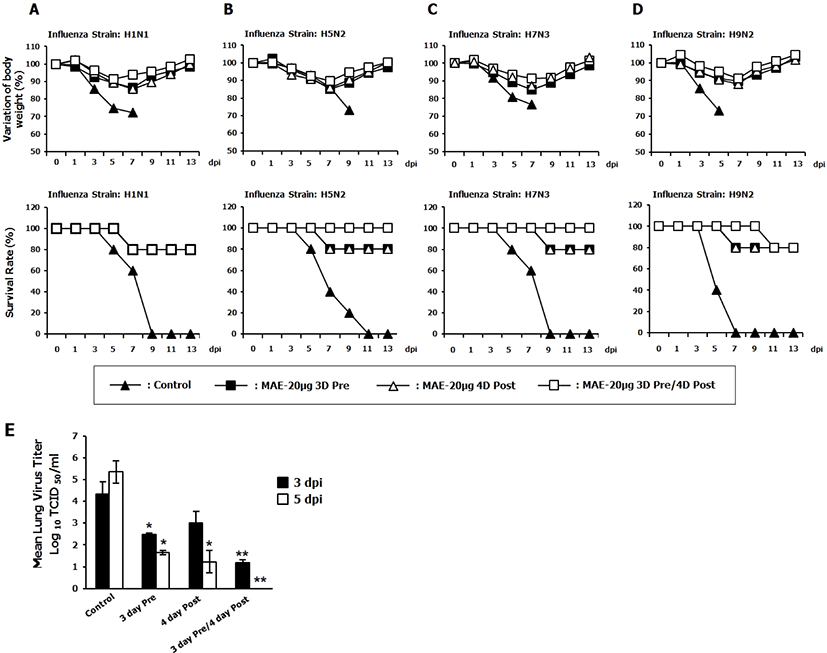
To further confirm the antiviral effect of MAE in-vivo, prophylactic and therapeutic effects of herb extract against influenza A virus infection was evaluated in a murine model of influenza A infection. BALB/c mice were infected with 5MLD50 of {A/PR/8/34(H1N1)}, {A/Aquatic bird/Korea/W81/2005(H5N2)}, {A/Aquatic bird/Korea/W44/ 2005(H7N3)}, and {A/Chicken/Korea/116/2004(H9N2)} influenza A subtypes. The mice were orally inoculated with MAE before and after infection with lethal doses of influenza A virus. The untreated (PBS) group showed significant body weight losses over the time. Moreover, the control group succumbed to death by 9 days after the challenge for all the viruses tested. In contrast, mice that received pre-treatment, post-treatment and both pre and post-treatment of MAE showed ≤ 20% body weight loss between 5 and 7 day post infection (dpi), commenced to recuperate their lost weight by 7 dpi, and returned to the normal status by 13 dpi (Fig. 3A–D). The herbal extract-treated groups had significantly higher survival rates compared with the PBS treated group. In addition, we determined the ability of MAE to inhibit the viral replication in the lungs of infected mice by performing the H1N1 challenge experiment, which help to confirm the above statement. Since mice from 5MLD50 H1N1 infected control group tend to die at 5 dpi and show severe disease symptoms in the early time of infection, mice were infected with 3MLD50 of the same strain of influenza virus for the lung virus titer determination. Mice from the control, pre-treated, post-treated and both pre and post-treated groups were euthanized and the lungs were collected aseptically at 3 days, and 5 days from the last post-inoculation (3 dpi and 5 dpi) for viral titration assay. Interestingly, Pre-treated, post-treated and both pre and post-treated mice had lower lung viral titers than that of the untreated group at 3 and 5 dpi (Fig. 3E). In summary, these results confirm that MAE has both prophylactic and therapeutic effects in vivo against different subtypes of influenza.
Discussion
Traditional medicine usage is becoming increasingly popular due to the drawbacks of conventional medicine, such as side effects, higher cost, and lack of efficacy [28]. Herbal or other traditional medicines have been used as remedies against infectious diseases over thousands of years because of their significant anti-inflammatory and anti-microbial activity, as well as low degree of adverse effects. In 2001 and 2002, nearly one-quarter of the global baseline drugs were natural products or derivatives of natural products [29]. Recent studies elaborate in China, their medicinal herbs account for 10% of prescription drugs [30]. A large number of global traditional herbs and their derived single components have shown a wide range of antimicrobial effects on various pathogens, as well as antioxidant, anti-inflammatory, antibacterial, antifungal, anti-plasmodial, and immune-modulatory effects [2]. Among the auspicious medicinal plants, Melia azedarach is a herb which has rich historical background and is mainly found in wild habitats in India, China, Korea, and most of the Arabic countries [31].
In the present study, we demonstrated the antiviral effect of MAE against a broad spectrum of viruses using in vitro and in vivo with diverse subtypes of the Influenza virus in mice model. We confirmed that the MAE strongly inhibited the viral replication in macrophages and epithelial cells in pre-treatment, co-treatment and post-treatment status (Fig. 1). Herbal extract markedly inhibited viral replication, and its effect was similar pattern to that of IFN-β treatment (positive control).
First, after viral infection, PRRs of the host senses the viral pathogen-associated molecular pattern (PAMPs) [32] and rapidly induces ISG and pro-inflammatory cytokine secretion for developing an innate antiviral immune response. The activation of the antiviral immune response at the early stage protect the host by controlling viral replication [32]. In this study, we found that the induction of antiviral immune response was remarkably increased after pre-treatment of MAE in cell lines. MAE show a strong ability to induce mRNA expression of the ISGs and a high potential to activate IFN-β and pro-inflammatory cytokine, such as IL-6 to induce protection against viral infection in cell lines. Next, to elucidate the described features of MAE in antiviral effects, we checked that MAE treatment induce the phosphorylation of IRF-3, STAT1, and TBK1 in a time-dependent manner, providing evidence for the downstream signal transduction in the type I IFN signaling pathway as well as the activation of the NF-κB pathway (p65). Consequently, MAE can activate the antiviral signaling molecules and lead to the production of Type I IFNs and proinflammatory cytokines that play an important role in stimulating the antiviral state and the subsequent suppression of virus replication.
Second, herbal medicines that are effective at the co-treatment phase can be divided into three stages based on their mode of antiviral mechanism, namely the virucidal effect, attachment inhibition, and penetration inhibition [33]. Co-treatment of MAE could significantly suppress the H3-GFP, EV71, and BRV virus replication than non-treated control. Consequently, it is possible that extracellular and intracellular activities of MAE against viruses could be attributed to its ability to bind and/or to inactivate important structural and/or non-structural protein(s) [34]. The final consequence of the post-treatment possibly correlates with the viral entry inhibition, cellular replication steps of virus or inhibition of viral particle release from the replicated cells [35, 36]. Several studies have been progressed to introduce components to be effective either on entry or replication processes of the virus in host [37–39]. According to the Shahsavandi et al., 2017 [36], the suppression of Influenza viral replication by post-treated medicinal herb, was due to inhibition of viral HA protein activities that involved in membrane fusion formation [36]. In the present study, post-treatment of MAE at 2, 4, and 6 hours post virus infection could significantly suppress the virus replication. Thus, the therapeutic effect of MAE probably could correlate with one or more of the aforementioned antiviral mechanisms. However, further studies are needed to identify the specific function behind MAE at the co-treatment and post-treatment stages.
Interestingly, oral administration of MAE increased the survival rate of BALB/c mice against 5MLD50 of influenza A subtypes including {A/PR/8/34(H1N1)}, {A/Aquaticbird/Korea/W81/2005(H5N2)}, {A/Aquaticbird/Korea/W44/2005(H7N3)}, and {A/Chicken/Korea/116/2004 (H9 N2)} (Fig. 3) and pre-treatment post-treatment and both pre and post treatment of MAE shows a remarkable reduction of lung virus titer than control which correlating with the previous result. These results suggest that MAE is strong enough to inhibit viral replication and promote the survival of mice against lethal infections by diverse subtypes of influenza A viruses. It is important to determine the cytotoxic concentration of the herbal extract in different cell lines and effective concentration against the different viruses in vitro, which could facilitate information regarding the safety margin of the herbal extract for therapeutic and prophylactic purposes. Investigation of cytotoxic concentration and effective concentration of MAE in different cell lines and different viruses respectively is in our future study plan. Previously reported phytochemical studies on Melia azedarach have identified more than 300 chemical compounds, such as hydroxycoumarin [40], β-carotene, tocopherol and saqulene [41] lignans [40, 42, 43], steroids [44], flavonoids (quercetin), steroids, terpenoids [45, 46]. Among them, relationship between antiviral effects and known or unknown component must be studied further. And also, to understand the mechanism of antiviral effects and immunomodulatory effects by Melia azedarach needed further studies.
In summary, the desirable safety margin and exhibited a broad spectrum of antiviral activity against RNA and DNA viruses, suggest that the aqueous extract of Melia azedarach may be a potential candidate for antiviral treatment against many virus diseases. Therefore, oral administration of Melia azedarach could have potential prophylactic or therapeutic applications in both human and livestock.