





Introduction
Lysyl oxidase (LOX) is a copper-dependent amine oxidase responsible for the post-translational modification of collagen and elastin fibrils by oxidizing ε-amino groups of peptidyl lysine residues to α-aminoadipic δ-semialdehydes groups in the extracellular matrix [1, 2]. In recent years, five members of the LOX family, LOX and LOXL1-4, have been identified. All these isoforms contain the characteristic C-terminal domains of the LOX family, such as a copper-binding domain, residues for lysyl-tyrosyl quinone (LTQ), and a cytokine receptor-like (CRL) domain [3–5]. Previously, we identified a novel LOX-variant 2 (LOX-v2) that contains the characteristic C-terminal domains of the LOX family but lacks the N-terminal propeptide region [6]. An alternative promoter element present in the exon 1 region of the LOX gene was required for the differential expression of LOX-v2, and LOX-v2 showed significant levels of amine oxidase activity toward collagen I and elastin [6]. Absence of the propeptide region in LOX-v2 suggests that this novel variant may be subject to distinct intracellular processing and then may localize in a cellular compartment different from where LOX resides.
In addition to the formation of lysine-mediated crosslinks in collagen and elastin fibrils, several novel functions of LOX have been revealed, including tumor suppression, cellular senescence, chemotaxis, and modification of histones [7–11]. Interestingly, several studies have reported that LOX is also present in nuclear locations in addition to the extracellular matrix [12–14]. Further, the nuclear histone H1 protein was oxidized by LOX [15], and histone H1 and H2 were shown to interact with LOX [16]. Taken together, these findings suggest that the LOX family proteins may play a novel functional role in the nucleus, beyond the amine oxidase activity toward collagen and elastin fibrils in the extracellular matrix.
In this study, we report that LOX-v2 localizes in the nucleoplasm with amine oxidase activity toward histone proteins. The transiently expressed LOX protein localized in the perinuclear, cytoplasmic, and extracellular areas, whereas the LOX-v2 protein predominantly localized in the nucleoplasm. LOX-v2 showed a significant level of amine oxidase activity toward histone H2A in in vitro peroxidase-coupled fluorometric assays. Our findings suggest that LOX-v2 localizes in a cellular location distinct from where LOX resides, playing a functional role different from that of LOX.
Materials and Methods
The expression plasmids of LOX and LOX-v2 were constructed as previously described [6]. Briefly, the cDNAs encoding the processed LOX (codons 169-417) and the full-length LOX-v2 were PCR-amplified from the cDNA of HEK293 cells using Pfu polymerase (Stratagene, La Jolla, CA, USA) according to the manufacturer’s suggestions. The PCR conditions consisted of 30 cycles at 94°C for 60 s, 56°C for 60 s, and 72°C for 60 s, with a predenaturation at 94°C for 4 min and a final extension at 72°C for 7 min. The PCR amplified DNA fragments were gel-purified and then subcloned into pET21a (Novagen, Madison, WI, USA) and pFLAG-CMV-5a (Sigma-Aldrich, St. Louis, MO, USA). The oligonucleotide primers used for constructions of pET21a-LOX, pET21a-LOX-v2, pFLAG-LOX, and pFLAG-LOX-v2 are listed in Table 1. Unique restriction sites, NheI or HindIII for pET21a and EcoRI, BamHI, or KpnI for pFLAG-CMV-5a, were introduced in the primers for convenient subcloning (Table 1). Sequence fidelity of all the constructs was confirmed by DNA-sequencing analysis, using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s protocol.
Purification and refolding of the LOX and LOX-v2 recombinant proteins were performed as we previously reported [17]. The pET21a-LOX and pET21a-LOX-v2 expression constructs were transformed into the Escherichia coli strain BL21. After incubation with 1 mM isopropyl-1-thio-b-D-galactopyranoside (IPTG) at 37°C, the transformants were harvested and lysed with a buffer containing 50 mM Tris, pH 8.0, 1 mM EDTA, 100 mM NaCl, 1 mM PMSF, and 1 mg/mL of lysozyme. During the inverted mixing, 1% Triton X-100, and 0.1 mg/mL DNase were added and then sonicated by 3 bursts of 10 seconds (amplitude 60). After centrifugation, the inclusion bodies were washed and solubilized in a buffer containing 6 M urea, 10 mM K2HPO4, pH 8.2, and 3 mM β-mercaptoethanol. The LOX and LOX-v2 recombinant proteins were then purified using the Ni-NTA agarose resins (Qiagen, Hilden, Germany) according to the manufacturer’s suggestion. The eluted LOX and LOX-v2 proteins were collected and refolded through stepwise dialysis in a buffer containing 10 mM K2HPO4, pH 9.6, 200 μM CuCl2, and 2% sodium N-lauroylsarcosinate and then in a buffer containing 10 mM K2HPO4, pH 9.6, and 5 μM CuCl2. The protein samples were further dialyzed in 10 mM K2HPO4, pH 9.6. The dialyzed proteins were lyophilized in the presence of 10 mM trehalose in a freeze dryer (Labconco, Kansas City, MO, USA). All purification and refolding procedures were performed at 4°C. The purity and sizes of the LOX and LOX-v2 proteins were determined by SDS–PAGE.
The amine oxidase activity of the LOX and LOX–v2 recombinant proteins was assessed by peroxidase-coupled fluorometric assays with the Amplex red hydrogen peroxide assay kit (Molecular Probes, Eugene, OR, USA) as previously described [18]. Each reaction contained 10 μg of the purified LOX or LOX-v2 recombinant protein in a reaction volume of 200 μL. The synthetic heteropolymer lysine/tyrosine (4:1) (20–50 kDa, Sigma-Aldrich), histone H2A (BPS Bioscience, San Diego, CA, USA), and collagen I (Sigma-Aldrich) were used as substrates at 5 ng/μL, 24 ng/μL, and 100 nM, respectively. Parallel assays were performed in the absence or presence of 1 mM β-aminopropionitrile (BAPN) for 1 h at 37°C. Fluorescence was measured using a fluorescence spectrophotometer (Molecular Devices, San Jose, CA, USA) with excitation and emission wavelengths of 500 and 650 nm, respectively. Specific amine oxidase activity was calculated by interpolation with fluorescence values from an H2O2 calibration curve and then expressed as nM of H2O2 produced per 10 μg of the LOX and LOX-v2 recombinant proteins.
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (GIBCO, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in 5% CO2. For transfection, 8.0 × 105 HEK293 cells were plated in gelatin coated cover slips in 6 well plates, grown for 18 h, and then transfected with a plasmid encoding LOX-FLAG or LOX-v2-FLAG, using the Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions. After 6 h of transfection, the medium containing the plasmid DNA complexes was replaced by the fresh medium containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. At post-transfection 48 h, cells were fixed in 4% paraformaldehyde for 20 min and then permeabilized with 0.2% Triton X-100 in PBS for 5 min. The cells were blocked with 3% goat serum for 30 min, washed with PBS, and then incubated anti-FLAG antibody (Abcam, Cambridge, MA, USA; 1:250). Staining was revealed by an Alexa Fluor 568-labeled secondary antibody (Invitrogen, 1:1,000). Nuclei were stained with 4’-6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific, Waltham, MA, USA; 1:1,000). Confocal images were scanned using the Fluoview FV1000 confocal laser scanning microscope (Olympus, Tokyo, Japan).
Results
In an attempt to produce a large amount of the LOX and LOX-v2 proteins for amine oxidase assays, the cDNAs corresponding to the processed 32 kDa form of LOX and the full-length LOX-v2 were subcloned into the pET21a vector in which the inserted LOX and LOX-v2 cDNAs were under control of the strong bacteriophage T7 promoter. The processed form of LOX starts at codon D169, and LOX-v2 uses codon M231 of LOX as the initiation codon. Both proteins contain the copper-binding domain, the LTQ residues, and the CRL domain in the C-terminus (Fig. 1A). After induction with 1 mM IPTG at 37°C, cell lysates were fractionated into different cellular compartments, such as cytoplasmic extracts, periplasmic extracts, and inclusion body fractions, and most of the recombinant proteins was expressed as an insoluble form in inclusion bodies (data not shown). The apparent sizes of the expressed recombinant proteins were in good agreement with the deduced molecular mass; 32 and 24 kDa for the LOX and LOX-v2 recombinant proteins, respectively. The LOX and LOX-v2 recombinant proteins were extracted with 8 M urea from the inclusion bodies and then purified using the Ni-NTA agarose column (Fig. 1 B and C).
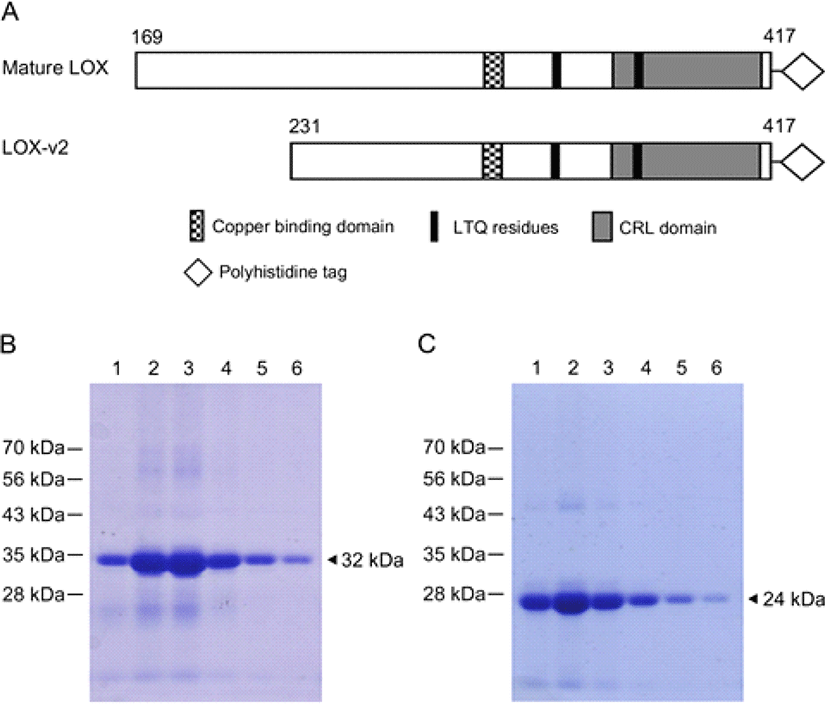
To refold the recombinant proteins denatured by urea during purification, we performed a dialysis in the presence of 2% sodium N-lauroylsarcosinate and 200 μM of CuCl2. Sequentially, we did second dialysis without any detergent but in the presence of 5 μM CuCl2. Finally, proteins samples were further dialyzed only in the potassium phosphate buffer. After purification, the hexa-histidine tagged LOX and LOX-v2 recombinant proteins were over 95% pure on SDS-PAGE gels (Fig. 1 B and C).
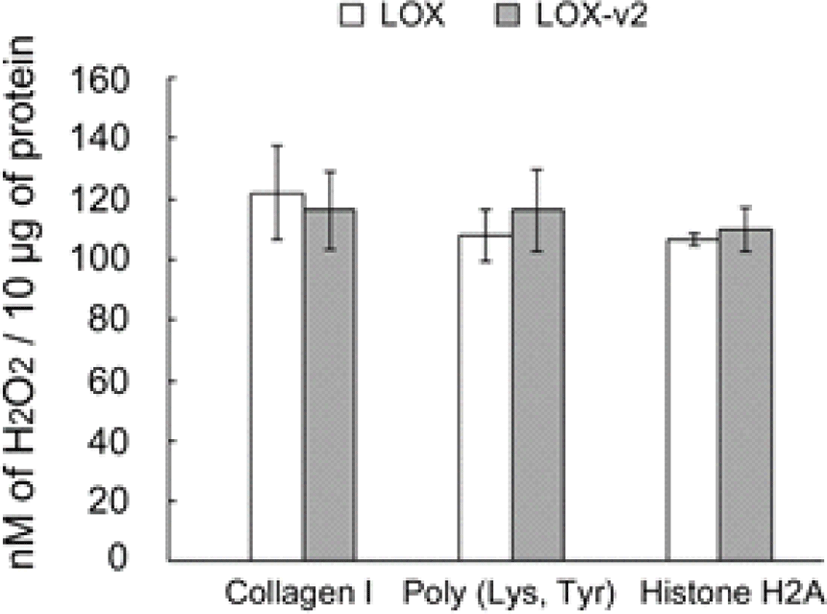
The purified and refolded LOX and LOX-v2 recombinant proteins were assessed for amine oxidase activity, using in vitro peroxidase-coupled fluorometric assays. To test the amine oxidase activity toward histone proteins, we used histone H2A as a substrate. Collagen I and a synthetic lysine/tyrosine heteropolymer were also tested as positive controls for the amine oxidase activity. Both the LOX and LOX-v2 recombinant proteins showed significant levels of amine oxidase activity toward all the substrates tested without significant differences in the activity (Fig. 2). The amine oxidase activities of LOX and LOX-v2 recombinant proteins were inhibited to the basal levels by BAPN, a well-known irreversible inhibitor of amine oxidase activity of the LOX family proteins (data not shown). Noticeably, both LOX and LOX-v2 showed significant levels of amine oxidase activity toward histone H2A, as comparable to the positive controls, collagen I and the lysine/tyrosine heteropolymer (Fig. 2). These results suggest that both LOX and LOX-v2 function as an amine oxidase toward the histone protein.
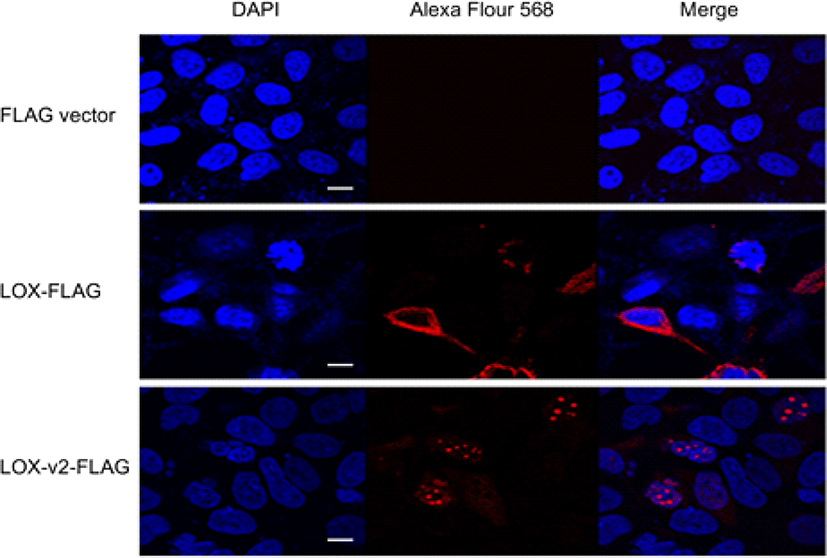
LOX-v2 uses the codon M231 of LOX as the initiation codon, sharing the same open reading frame with LOX. Thus, it is not possible to generate an antibody that specifically distinguishes LOX-v2 from LOX. To determine the cellular location of LOX and LOX-v2, we designed recombinant constructs with a FLAG epitope at the C-terminus of LOX and LOX-v2. The recombinant constructs were transiently expressed in HEK293 cells, and then the cellular localization was monitored by immunofluorescence with an antibody against the FLAG epitope. In immunofluorescence staining, LOX-v2 was noticeably stained in the nucleus, showing a punctate pattern in the nucleoplasm (Fig. 3). By contrast, LOX was observed in the perinuclear, cytoplasmic, and extracellular areas (Fig. 3).
Discussion
The LOX precursor is synthesized as a 48 kDa precursor protein in the cytoplasm and goes through post-translational modifications throughout the secretion pathway to the extracellular matrix [19]. After secretion into the extracellular matrix space, the N-terminal propeptide region of 147 amino acids is cleaved off between residues G168 and D169, resulting in an enzymatically active 32 kDa form of 249 amino acids [19]. Previously, LOX was reported as a ras recision gene in the phenotypic reversion of ras-transformed cells [7]. Further, LOX downregulation was reported in prostate, bronchogenic, and gastric carcinomas [20–22]. However, upregulation of LOX was detected in metastatic cells of breast carcinomas [23], suggesting bifunctional roles of LOX in the suppression and progression of tumorigenesis. The propeptide region of LOX was reported to diminish the fibroblast growth factor-2-induced proliferation in prostate cancer cells [24] while the processed 32 kDa form of LOX was shown to induce the hypoxia-induced metastasis of breast carcinomas [25]. These findings suggest that the tumor suppression activity may reside in the propeptide region, whereas the processed 32 kDa form of LOX may be more likely involved in the invasiveness of metastatic cancer cells. The absence of the propeptide region in LOX-v2, thus, suggests that the tumor progression activity of LOX in metastatic cancer cells may be derived, at least in part, by LOX-v2. The non-existence of the propeptide region in LOX-v2 further suggests that LOX-v2 may be subject to intracellular processing different from what LOX undergoes and then localizes in cellular compartments distinct from where LOX is located.
Previously, a 24 kDa protein band immune-reactive to the LOX antibody was detected in the purified fractions of bovine aorta [26], and 25–28 kDa sizes of LOX were detected in the nuclear fraction of rat aorta smooth muscle cells [13], suggesting that the smaller sizes of LOX ranging from 24 to 28 kDa may be resulted from the expression of LOX-v2. LOX is an extracellular enzyme that catalyzes the crosslink formation in collagen and elastin fibrils, however, LOX was reported to be also present in the nuclear compartments [13]. The nuclear localization of LOX has been further reported in several different tumor tissues, including clear cell renal cell carcinoma, rectal carcinoma, and high-grade serous ovarian carcinoma [27–29]. In our findings, both LOX and LOX-v2 showed significant amine oxidase activity toward histone H2A in in vitro peroxidase-coupled fluorometric assays, however, only LOX-v2 displayed cellular localization in the nucleus where histone proteins are located. These results, thus, suggest that LOX-v2 may be a regulatory enzyme responsible for the post-translational modification of histones in the nucleus. Further studies on the specific subnuclear locations of LOX-v2, along with further detailed analysis on substrate specificity of LOX-v2, will be critical for determining the specific functional roles of LOX-v2 in the nucleus.