




Introduction
Stroke due to sudden interruption of blood flow to the brain carries a high risk of death and is the main cause of serious long-term disability. Deprivation of oxygen supply to the brain tissue due to the reduction of blood flow leads to activation of the ischemic cascade with activation of a series of molecular mechanisms [1, 2]. The major pathological mechanisms of ischemic brain injury include excitotoxicity, oxidative stress, inflammation, and apoptosis, which are associated with mitochondrial dysfunction and a rapid decrease of ATP [3]. With energy depletion, disrupted membrane potential causes massive neuronal depolarization and an increase of intracellular Ca2+ concentration ([Ca2+]i), resulting in an extracellular buildup of excitatory amino acids and consequent excitotoxicity [4, 5].
Vitis, a kind of grapes, has various pharmacological effects including protection against fatty liver, hypertension, cardiac diseases and arthritis [6]. Vitis amurensis contains a large number of polyphenolic compounds including resveratrol possessing potent anti-oxidant and anti-inflammatory properties [6, 7]. Aralia belonging to the family Araliaceae has long been recognized as a therapeutic herb for its antinociceptive, antidiabetic, antioxidant and anti-inflammatory activities in China, Japan and Korea. Diterpenes isolated from the root of Aralia cordata were known as active constituents for analgesic and anti-inflammatory agents [8, 9]. Studies have indicated that extracts from the aerial parts of this plant inhibit cyclo- oxygenase (COX)-1, COX-2, and COX-2-dependent prostaglandin E2 (PGE2) generation, as well as hyperalgesia during peripheral inflammation [10-12]. Glycyrrhiza species (Leguminosae) have long been recognized as medicinal plants in traditional oriental medicine, and are widely used due to their diverse pharmacological properties including vasorelaxant [13], anti-inflammatory [14], and anti-oxidant effects [15]. There is growing evidence for Glycyrrhizae radix, the root of Glycyrrhiza uralensis, with beneficial effects in brains such as anti-depressant [16] and memory enhancing effects [17] and protection against ischemic brain damage in animal models [18].
In our previous studies, aerial parts, leaves and stems, of Vitis amurensis and Aralia cordata showed a neuroprotective effect against ischemic brain damage and β-amyloid protein-induced memory deficits in animals, and oxidative and excitotoxic neurotoxicity in cultured neurons [19-22]. Glycyrrhizae radix also showed neuroprotection against β-amyloid protein-induced neurotoxicity in cultured neurons [23]. In a recent study, we demonstrated that an ethanol extract of a mixture of Vitis amurensis, Aralia cordata, and Glycyrrhizae radix, named SSB, protected against β-amyloid protein-induced memory deficit in mice and cultured neuronal cell death [24]. The current study was conducted to further verify the neuroprotective effect of SSB on cerebral ischemic injury and excitotoxicity using both in vivo and in vitro studies. We examined the protective effect of SSB against transient focal ischemic brain damage induced by middle cerebral artery occlusion for 2 hr followed by reperfusion for 24 hr (MCAO/reperfusion) in rats and against excitotoxicity induced by glutamate in cultured rat cortical neurons.
Materials and Methods
The leaves and stems of Vitis amurensis and the aerial parts of Aralia cordata were collected at Keryong Mountain in Daejeon, Korea, from May to July, and Glycyrrhizae radix (Korean) was purchased from Daegu Oriental Pharm Co., Daegu, Korea. These plants were identified by Dr. Kyung-Sik Song, Kyungpook National University. From each of the three plants, 200 g was mixed, extracted with ethanol (3 L × 24 hr × 3) at room temperature, filtered, and concentrated under reduced pressure using a rotary evaporator to yield an ethanol extract (SSB, 64 g) , which was stored at room temperature until required. Glutamate, 3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyl-tetrazolium bromide (MTT), 2,3,5-triphenyltetrazolium chloride (TTC), Dulbecco's modified Eagle's medium (DMEM), Joklik-modified MEM, and poly-Llysine were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Fluo-4 AM and 2',7'-dichlorodihydrofluorescein diacetate (H2DCF-DA) were purchased from Molecular Probes Inc. (Eugene, OR, USA). Fetal bovine serum was purchased from JRH Biosciences (Lenexa, KS, USA).
Sprague-Dawley (SD) rats and pregnant SD rats for cerebral neuron culture were purchased from Daehan Bio- Link Co., Ltd. (Chungbuk, Korea). Animals were housed in an environmentally controlled room at 22 ± 2 °C, with a relative humidity of 55 ± 5%, a 12 hr light/dark cycle, and food and water ad libitum. The procedures involving experimental animals complied with the animal care guide lines of the National Institutes of Health and the Animal Ethics Committee of Chungbuk National University.
Before surgery, male SD rats weighing 280~300g were fasted overnight with free access to water. Focal cerebral ischemia was induced by MCAO for 2 hr followed by reperfusion for 24 hr as previously described [25]. Following evaluation of neurological score after MCAO/ reperfusion, the rats were sacrificed by decapitation under anesthesia (diethyl ether), and brains were rapidly removed. The infarct and edema volumes of brain tissue were measured using TTC staining [25]. The infarct area in each slice was calculated by subtracting the normal ipsilateral area from that of the contralateral hemisphere to reduce errors due to cerebral edema. The edema volume was calculated by subtracting the contralateral hemisphere volume from the ipsilateral hemisphere volume. These were presented as a percentage relative to those of the contralateral hemisphere [25]. Neurological status of animals was evaluated using the Menzies method [26]. SSB (5, 10, and 25 mg/kg) suspended in distilled water was administered orally three times: 0.5 hr before and 1 hr after MCAO, and 1 hr after reperfusion.
Primary cortical neuron cultures were prepared using SD rat fetuses on embryonic days 15 to 16 as previously described [25]. Neurotoxicity experiments were performed on neurons after 5~6 days in vitro. Cultured neurons were treated with 500 μM glutamate in a HEPESbuffer (incubation buffer, pH 7.4) containing 8.6 mM HEPES, 154 mM NaCl, 5.6 mM KCl, and 2.3 mM CaCl2 at 37°C for 12 hr (unless otherwise indicated) to induce neurotoxicity. SSB was dissolved in dimethyl-sulfoxide (DMSO) at concentrations of 50 mg/mL and further diluted in the incubation buffer. The final concentration of DMSO was less than 0.1%, which did not affect cell viability. The neurons were treated with SSB 20 min prior to treatment with 500 μM glutamate. Neuronal viability was measured using the colorimetric MTT assay [25]. Changes in [Ca2+]i and generation of reactive oxygen species (ROS) were measured with fluorescent dyes, fluo-4 AM and H2DCF-DA, respectively, using a laser scanning confocal microscope (TCS SP2 AOBS; Leica, Heidelberg, Germany) [25].
Results
Representative brain slices from vehicle- and SSBtreated groups are shown in Fig. 1A. After MCAO/ reperfusion, a large ipsilateral cerebral infarction was observed in the rat brain. Normal brain tissue stained deep red by a reaction between TTC and mitochondrial dehydrogenase, whereas the infarct tissues did not stain. Orally administered SSB (10 and 25 mg/kg) significantly reduced the infarct formation showing 19.3 ± 3.0% infarction by 25 mg/kg SSB compared to that of vehicletreated control rats (45.9 ± 2.8%). The edema volume increased by MCAO/reperfusion was also significantly reduced by SSB (25 mg/kg) compared to that of vehicle treated groups (Fig. 1B). Animal body temperatures were monitored for 6 hr after cerebral reperfusion commenced, and no significant differences were observed between the SSB and control groups (data not shown). Thus, the observed neuroprotective effect of SSB could not be attributable to hypothermia.
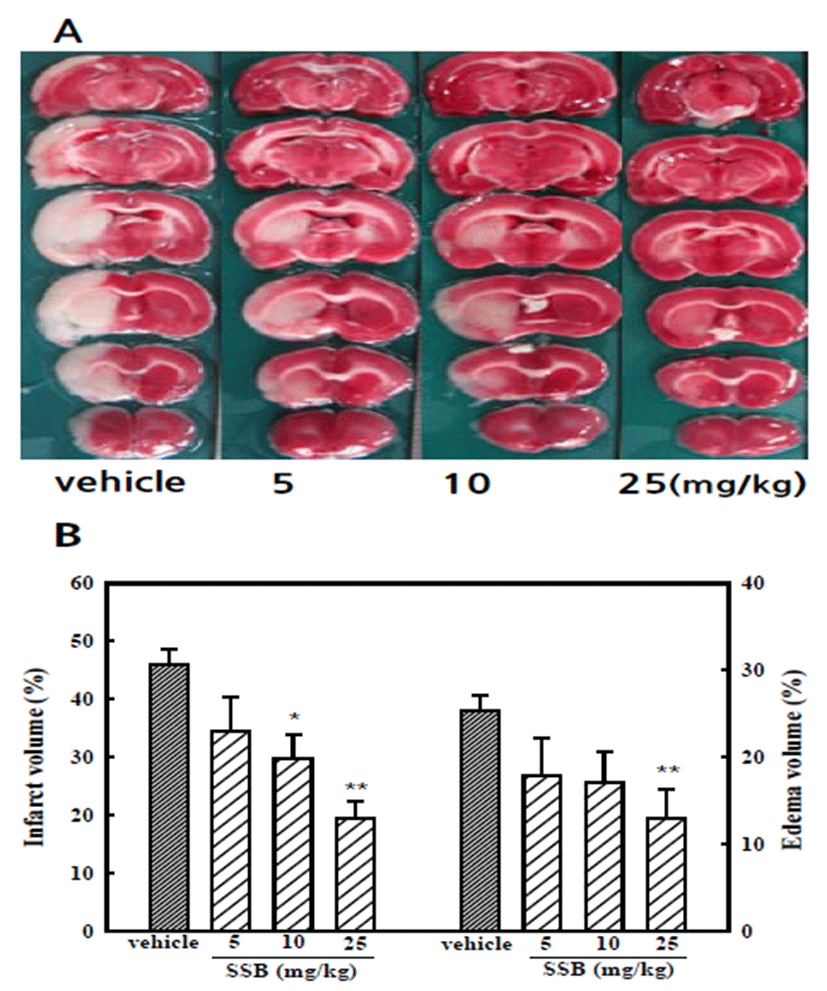
MCAO/reperfusion rats showed neurological deficits, including circling movement and decreased grip of the contralateral forelimb. The neurological deficit score was reduced significantly to 1.9 ± 0.3 in the SSB-treated (25 mg/kg) group compared to that of the vehicle-treated control group, which scored 3.6 ± 0.2 (Table 1).
| Group | Dose (mg/kg) | Neurological score |
|---|---|---|
| Vehicle | - | 3.6 ± 0.2 |
| SSB | 5 | 2.5 ± 0.2 |
| SSB | 10 | 2.2 ± 0.3* |
| SSB | 25 | 1.9 ± 0.3** |
After MCAO/reperfusion, neurological status of the animals used in Fig. 1 was evaluated. Five categories of neurological finding according to Menzies were scored: 0, no apparent deficits; 1, contralateral forelimb flection; 2, decreased grip of contralateral forelimb; 3, contralateral circling if pulled by tail; and 4, spontaneous contralateral circling. Values are expressed as mean ± S.E.M.
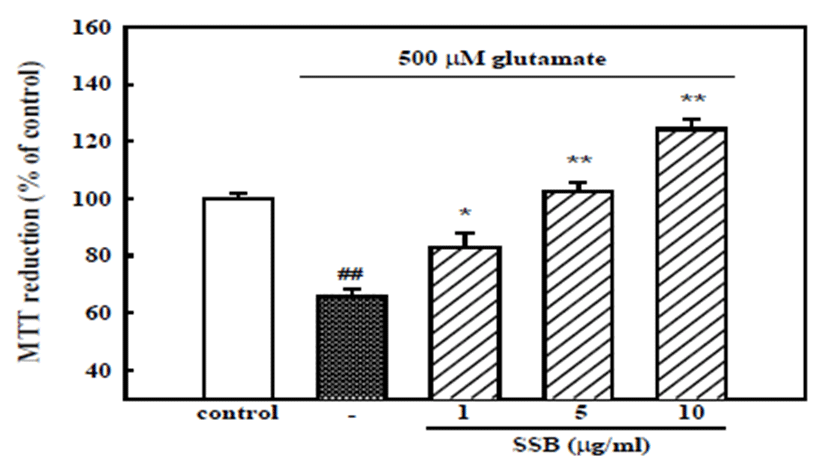
To elucidate the neuroprotective mechanism, the effect of SSB on neuronal damage induced by glutamate, which can be released from ischemic tissues to cause neuronal excitotoxicity, was examined. When cortical neurons were exposed to 500 μM glutamate for 12 hr, absorbance in the MTT assay was 66.0 ± 2.6% of that of the untreated control, indicating that glutamate caused neuronal death. Pretreatment of cortical neurons with SSB resulted in concentration-dependent inhibition of neuronal death induced by 500 μM glutamate showing cell viability of 102.3 ± 3.6 and 124.2 ± 3.2% with 5 and 10 μg/ml SSB, respectively (Fig. 2). This result suggests that SSB could also prevent neuronal cell death caused by serum-, glucose-, and Mg2+-free stress condition, since neurons incubated in the incubation buffer for 12 hr showed MTT absorbance of 83.1 ± 3.0% compared to that of neurons incubated in the normal growth medium (data not shown).
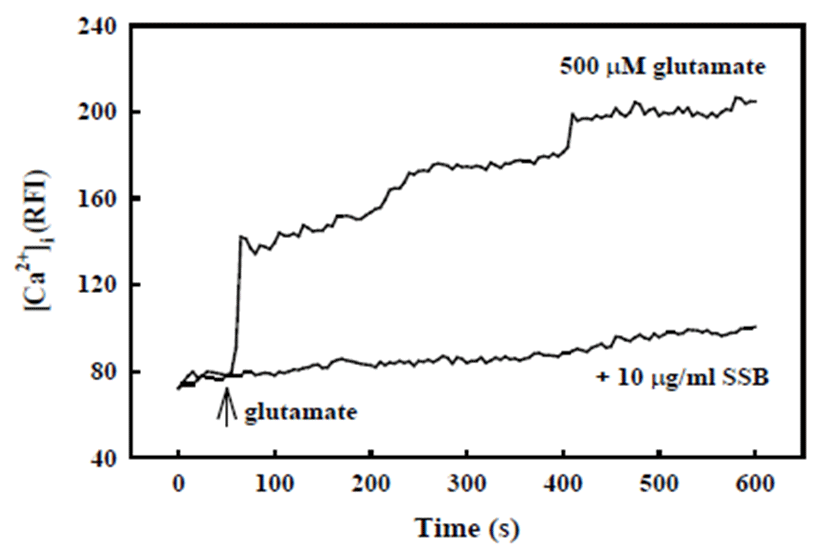
Glutamate-induced neuronal death is usually associated with the elevation of [Ca2+]i following N-methyl-D-aspartate (NMDA)-type glutamate receptor activation. As shown in Fig. 3, [Ca2+]i was rapidly elevated in response to treatment with 500 μM glutamate, which was maintained over 10 min. In contrast, SSB (10 μg/ml) significantly inhibited glutamate-induced increase of [Ca2+]i.
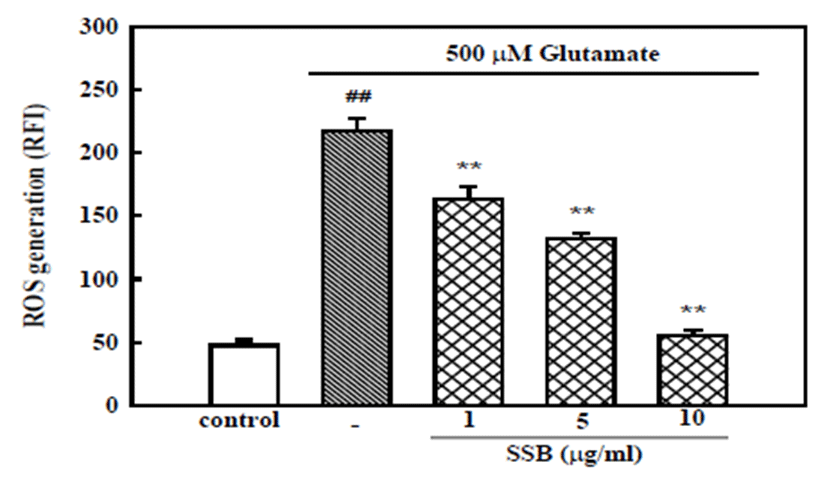
The involvement of oxidative stress in glutamate toxicity was examined by measurement of ROS accumulation after exposure of neurons to glutamate for 6 hr. In H2DCF-DA-loaded cortical neurons, 500 μM glutamate increased the fluorescence intensity, indicating that ROS were generated. In neurons treated with 500 μM glutamate, the relative fluorescence increased ~ 4.5-fold to 217.1 ± 4.2, compared to the value in control neurons (47.6 ± 10.6). SSB (1, 5, and 10 μg/ml) showed concentration- dependent inhibition of the glutamate-induced ROS generation (Fig. 4).
Discussion
The current study demonstrated the neuroprotective effect of SSB on transient focal cerebral ischemia-induced brain damage using an MCAO/reperfusion model in rats as well as glutamate-induced neurotoxicity in cultured cortical neurons. The focal cerebral ischemia model with transient MCAO followed by reperfusion in experimental animals, which was used to induce ischemic injury in the current study, has generally been accepted as the most appropriate model for human stroke [27]. Ischemia leads to the release of cellular toxic mediators and increases the permeability of the blood brain barrier (BBB) [28]. Hyperpermeability of the BBB leads to brain cellular swelling and causes brain infarction and edema [29, 30]. The size of brain infarct and edema correlates with severity of the neurological deficits [31]. MCAO/reperfusion produced significant impairments in neurological function, and brain infarction and edema were observed in the present study. However, SSB dose-dependently inhibited MCAO/reperfusion-induced brain infarct and edema formation, and neurological deficits.
Glutamate is a major excitatory neurotransmitter in the central nervous system (CNS), but excessive exposure to glutamate may cause neuronal dysfunction and even degeneration. Excitotoxicity through overactivation of NMDA receptors by the extensive release of glutamate is well established as an important trigger of tissue damage in focal cerebral ischemia [32, 33]. Our data also showed that MCAO/reperfusion-induced infarct and edema were blocked by the NMDA receptor antagonist MK-801 [20, 25]. Activation of NMDA receptors elevates the influx of Ca2+, and that of non-NMDA glutamate receptors promotes the influx of Na+, both of which can lead to mem- brane depolarization. In turn, depolarization can activate plasma membrane voltage-dependent Ca2+ channels, leading to additional Ca2+ influx [33, 34]. Furthermore, increased [Ca2+]i activates enzymes, such as xanthine oxidase and nitric oxide synthase (NOS), which are involved in ROS generation, leading to lipid peroxidation and neuronal damage [35, 36]. In the current study using cultured cortical neurons, glutamate induced neuronal cell death. Glutamate also increased [Ca2+]i and ROS generation. Treatment of neurons with SSB significantly suppressed glutamate-induced neuronal cell death, [Ca2+]i increase, and ROS generation in cultured cortical neurons. Therefore, it might be concluded that the favorable effects of SSB on MCAO/reperfusion-induced brain damage can be attributed to blockade of the apoptotic cascade after inhibition of [Ca2+]i increase and ROS generation, which were primarily triggered by excessively released glutamate under ischemic conditions.
Phytochemically, we isolated 11 resveratrol derivatives and resveratrol oligomers possessing anti-oxidant and anti-inflammatory activities from the leaf and stem of Vitis amurensis [37, 38]. Resveratrol derivatives have been demonstrated to inhibit Ca2+ entry through Ca2+ channels and to possess ROS scavenging activity [25, 39]. (+) Ampelopsin A, γ-2-viniferin, and trans-ε-viniferin isolated from the leaf and stem of Vitis amurensis inhibited glutamate-induced increases of [Ca2+]i, ROS generation, and resultant apoptotic neuronal death in cultured neurons in a previous work [21]. We have isolated oleanolic acid from Aralia cordata as one of the principle components for the inhibition of ischemic brain damage. Oleanolic acid inhibited hydrogen peroxide-induced neuronal death, and increases of [Ca2+]i, ROS generation [20]. Isoliquiritigenin isolated from Glycyrrhizae radix as an active component inhibited β-amyloid protein-induced neuronal apoptotic death, elevation of [Ca2+]i, and ROS generation [22]. Therefore, the primary inhibition of [Ca2+]i elevation by these active compounds contained in these three plants are probably involved in the mechanisms of anti-oxidative and additional neuroprotective effects of SSB against MCAO/reperfusion-induced brain injury. The mechanism by which these compounds block glutamate-induced [Ca2+]i elevation is unclear. The compounds may stabilize membranes in a manner that blocks Ca2+ influx via NMDA receptor-coupled voltage dependent Ca2+ channels. Supporting this hypothesis, studies demonstrated that resveratrols reduced NMDA and kainate sensitive excitatory currents in hippocampal neurons and L-type Ca2+ channel currents in rat ventricular myocytes [40, 41].
In previous reports, Vitis amurensis [21] and Aralia cordata [20] at the dosage of 100 and 50 mg/kg, respectively, induced approximately 50% neuroprotection against ischemic brain damage, while a mixture of three plants, SSB, showed a similar neuroprotection by a dosage of 25 mg/kg in the current study. This result indicates that three medicinal plants acted synergistically to produce neuroprotection, since maximal effect of SSB was shown at a much lower concentration than the sum of maximal effect concentration of each plant.
In conclusion, the current study demonstrated that SSB exhibits a neuroprotective effect against MCAO/reperfusion- induced cerebral ischemic damage. It might be concluded that this neuroprotective effect is related to the anti-excitotoxic action of SSB, and that SSB might be effectively applied for the treatment of ischemic stroke.