




Introduction
Bone is dynamic tissue that continuously synthesizes and destructs its own matrix by osteoblasts and osteoclasts, respectively [1, 2]. Bone resorption by osteoclasts is majorly regulated by two cytokines, macrophage/monocyte-colony forming factor (M-CSF) and receptor activator of nuclear factor kappa B ligand (RANKL) [3, 4]. Osteoclasts are differentiated from hematopoietic stem cells, particularly monocyte and macrophage lineage cells [3]. Binding of RANKL to its receptor, RANK, is known to trigger osteoclastogenesis [3]. RANK mainly activates mitogen-activated protein kinases (MAPKs; ERK, p38, and JNK) and nuclear factor kappa-B (NFκB) [5, 6]. In addition, calcium-mediated calmodulin-dependent kinase (CaMK) signaling is one of the most critical signaling pathways for inducing nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) expression through c-Fos, Fra1, Fra2, and cAMP-response element (CRE)-binding protein (CREB) [7, 8]. Cyclic adenosine 3’, 5’-monophosphate (cAMP) has been shown to induce target gene expression through activation of cAMP-dependent protein kinase (PKA) [9]. Up-regulation of the intracellular cAMP level can stimulate PKA-mediated CREB phosphorylation [10]. In addition, cAMP can evoke CREB phosphorylation through the p38-mediated mitogen- and stress-activated kinase 1 (MSK1) pathway [10].
MSK1 is a serine/threonine kinase that is stimulated by multiple extracellular molecules such as epidermal growth factor, TPA, and UV [11]. As up-stream kinases, PKA and MAPKs stimulate MSK1 phosphorylation. MSK1 has two kinase domains in its C- and N-terminal regions [11]. Further, Ser 360 and Thr 581 in MSK1 are phosphorylation sites for ERK and p38 [12]. Additional auto-phosphorylation sites in MSK1 include Ser 212, Ser 376, Ser 381, Ser 750, Ser 752, and Ser 758 [9]. MSK1 can also activate various downstream transcriptional factors, including CREB, ATF1, ATF2, p65, and STAT3 [11, 13, 14]. Although activation triggers and down-stream signaling pathways of MSK1 have been investigated, RANKL-induced MSK1 phosphorylation is not well known. In the present study, we demonstrated that MSK1 (Ser 376) was significantly phosphorylated by RANKL and might play a role in NFATc1 induction through CREB and c-Fos.
Materials and Methods
Recombinant M-CSF and RANKL were from Peprotech (London, UK). Antibodies against pMSK1 (Ser376), ATF2, pCREB (Ser133), and c-Fos were from Cell Signaling Technology (Cambridge, MA, USA). Anti- NFATc1 was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-β-actin was from Sigma-Aldrich (St. Louis, MO, USA). H89, SB203580, U0126, and PD98059 were obtained from Calbiochem (San Diego, CA, USA). CCK assay was from Dojindo (Kumamoto, Japan). Lipofectamine 2000 was from Invitrogen (Carlsbad, CA, USA). Luciferase system was from Promega (Madison, WI).
Bone marrow-derived macrophages (BMMs) were prepared as described previously [15]. For RANKL stimulation, BMMs were serum-starved for 5 hours. RANKL (500 ng/mL or an increasing dose) were applied to serum- free media for the indicated time. For signaling inhibitor experiments, BMMs were pre-treated with each inhibitor for the last 30 min of the serum starvation period and stimulated with RANKL (500 ng/mL) in the presence of inhibitors.
Cells were lysed with whole cell lysis buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS] with proteinase and phosphatase inhibitors. Cell lysates were subjected to Western blotting as described previously [15].
Bone marrow macrophages were cultured with M-CSF (30 ng/mL) plus RANKL (150 ng/mL) in the presence of increasing doses of H89. After 24 hours of incubation, 10% CCK solution was added to the cell culture media. The absorbance at 450 nm was detected by an ELISA reader after 30 min of incubation.
RAW264.7 cells were transfected with c-Fos luciferase construct in serum-free DMEM. After culturing with DMEM containing 10% FBS for 24 hours, cells were incubated with SB203580 (1 μM and 10 μM) in the absence or presence of RANKL for another 12 hours. Cells were then lysed with Glo lysis buffer. Luciferase activity was detected using a FLUOstar OPTIMA luminometer from BMG Labtech (Offenburg, German), after which the concentration of protein in each sample was normalized.
Recombinant retroviral vectors harboring a dominant negative form of CREB were kindly provided by Dr. Takayanagi H. from Tokyo Medical and Dental University. Plat-E cells were used for virus packaging, and infection procedure was performed as described previously [6].
Results
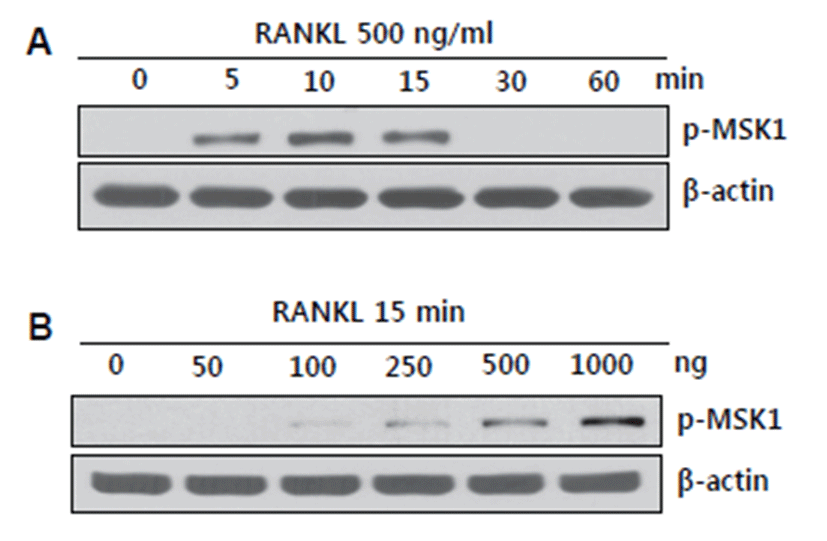
BMMs started to undergo differentiation into osteoclasts upon RANKL stimulation [6]. RANKL has been shown to activate various signaling pathways, including TRAF-mediated MAPKs, NFκB, and calcium-mediated CamK signaling [6, 16]. MSK1 is a known downstream effector molecule of PKA and MAPKs but has been rarely investigated regarding its role in RANKL-stimulated osteoclast precursor cells, especially BMMs. Although MSK1 phosphorylation at Thr 581 has been shown to play a role in bone destruction under RANKL-stimulated conditions [16], we instead focused on phosphorylation of MSK1 at Ser 376 by RANKL in BMMs. MSK1 activation was initiated at 5 min after RANKL stimulation and was sparsely detected after 30 min (Fig. 1A). When BMMs were stimulated with an increasing dose of RANKL for 15 min, MSK1 phosphorylation at Ser 376 was detected at the lowest dose (100 ng/mL of RANKL). Activation of MSK1 occurred in a dose-dependent manner (Fig. 1B). Since MSK1 phosphorylation at Ser 376 was shown to be required for kinase activity together with Thr 581 phosphorylation, RANKL-mediated activation of MSK1 at Ser 376 might regulate N-terminal auto-phosphorylation of MSK1 [12].
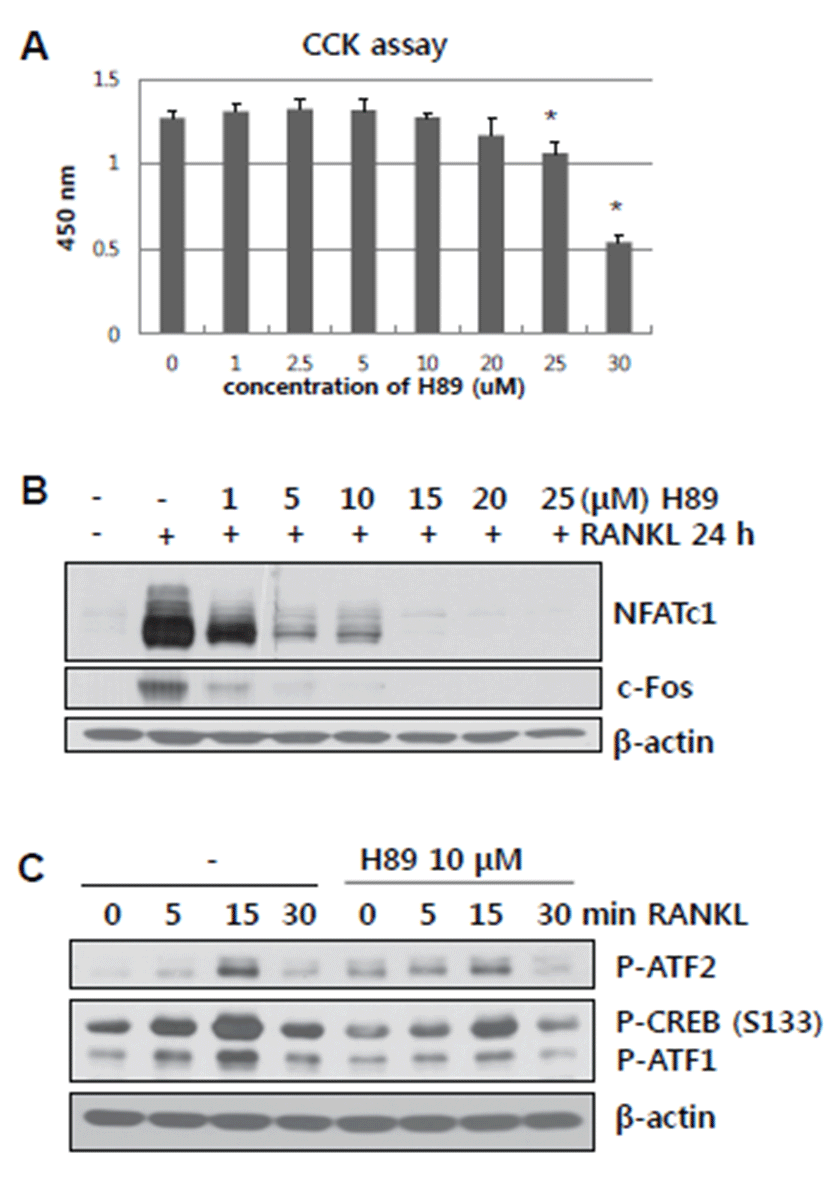
PKA is known to be activated by intracellular cAMP [9]. In addition, PKA directly stimulates CREB phosphorylation and indirectly mediates p38/MSK1 signaling [10]. Thus, we determined whether or not blockade of PKA by the chemical inhibitor H89 affects RANKL-mediated NFATc1 and c-Fos activation of ATF2 and CREB (Ser 133). An increasing dose of H89 was examined for its toxicity by CCK assay. The maximum dose of H89 used in this study was 20 μM. BMMs treated with H89 in the presence of RANKL for 24 hours showed significantly reduced RANKL-induced NFATc1 and c-Fos expression (Fig. 2B). Moreover, H89-treated BMMs showed low levels of RANKL-stimulated ATF2 and CREB (Ser 133) phosphorylation (Fig. 2C). PKA, which acts upstream of MSK1, participated in RANKL-induced NFATc1 and c-Fos expression by regulation of ATF2 and CREB activation.
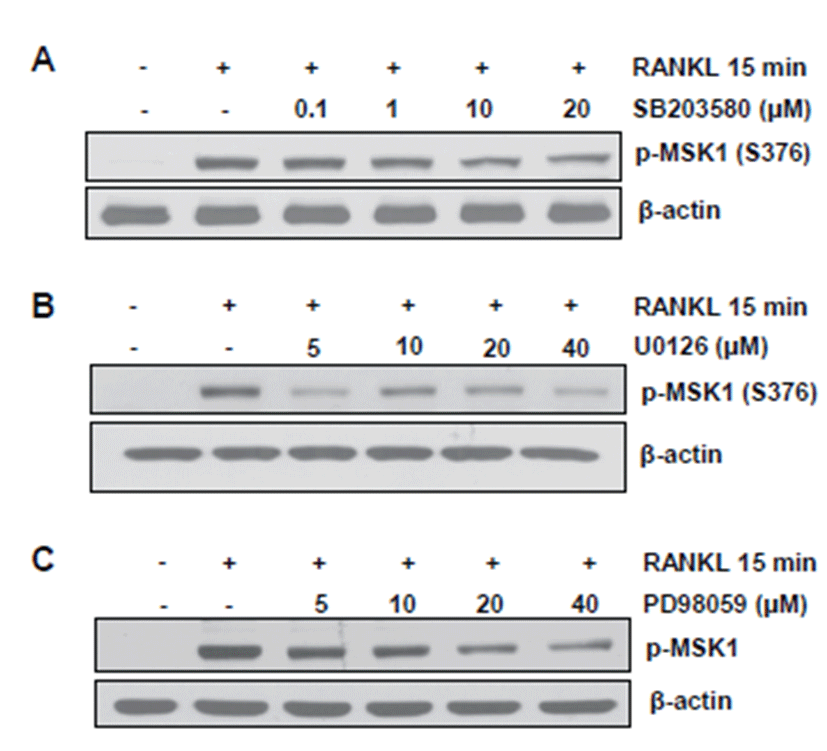
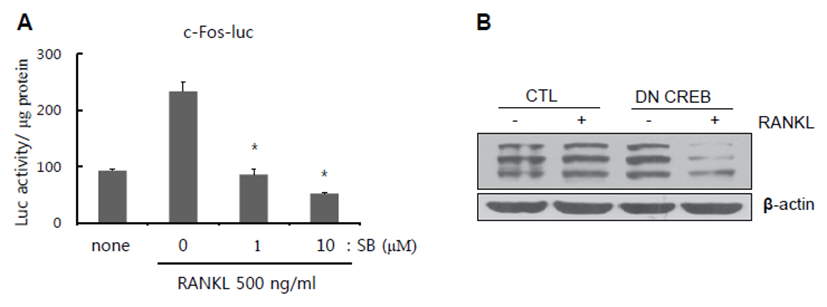
MAPK can act as an upstream kinase of MSK1 [12]. Thus, we investigated whether or not RANKL-induced MSK1 phosphorylation at Ser 376 is mediated by MAPK activation. For this, serum-deprived BMMs were stimulated with RANKL in the presence of increasing doses of p38 and ERK inhibitors. As shown in Figs. 3A-3C, phosphorylation of MSK1 at Ser 376 was suppressed by inhibitors. Next, to determine whether or not the final transcription factor in MSK1 signaling is c-Fos, we performed reporter assay with c-Fos luciferase vector. Since p38 was shown to affect MSK1 phosphorylation, we used p38 inhibitor to block p38-mediated MSK1 signaling. In our results, RANKL-induced luciferase activity was significantly reduced by treatment with p38 inhibitor (Fig. 4A). CREB is a crucial transcription factor for NFATc1 induction upon RANKL stimulation. In addition, we observed that CREB activation increased RANKL stimulation and was inhibited by H89 treatment. Therefore, we examined whether or not the dominant negative form of CREB (DN-CREB) could abolish NFATc1 induction by RANKL stimulation. As shown in Fig. 4B, NFATc1 induction was reduced by DN-CREB.
Discussion
RANKL is a crucial cytokine that can promote osteoclast differentiation [6]. As another crucial cytokine for osteoclast differentiation, M-CSF promotes expression of RANK, the receptor for RANKL, in osteoclast precursors such as BMMs [17]. Therefore, binding of RANKL to RANK initiates the differentiation of osteoclast precursors into mature osteoclasts [3]. RANKL-RANK signaling provokes various intracellular signaling molecules such as MAPKs, NKκB, and CaMK, all of which mediate induction of c-Fos and NFATc1, which are key transcriptional factors of osteoclastogenesis [6, 9]. In addition, CREB is known to induce NFATc1 expression. CREB is phosphorylated at Ser 133 by PKA, which promotes recruitment of co-activator proteins CBP and p300. MSK1 activates CREB phosphorylation at Ser 133 in response to activation of MAPK signaling, although MSK1 does not promote strong recruitment of CBP or p300 [18]. In this regard, MSK1 acts as a downstream signaling molecule of both PKA and MAPK. In the present study, we showed that c-Fos and NFATc1 induction by RANKL was significantly suppressed by PKA inhibitor. In addition, activation of MSK1 was modulated by MAPKs, especially p38 and ERK. Finally, c-Fos transcriptional activity was regulated by p38 inhibitor, and the dominant negative form of CREB suppressed NFATc1 induction by RANKL.
MSK1 is phosphorylated by active ERK and p38 to promote kinase catalytic activity in response to multiple stimuli [12]. MSK1 then phosphorylates downstream substrates such as CREB, ATF1, and p65 subunit of NFκB as well as histone H3 [19, 20]. MAPK-mediated MSK1 phosphorylation occurs at Ser 360 and Thr 581 [21]. A previous report detected activation of MSK1 at Thr 581 by RANKL stimulation in osteoclast precursors, and MSK1 regulates osteoclastogenesis together with bone destruction effect in vivo [16]. In the present study, we have for the first time observed MSK1 phosphorylation at Ser 376 upon RANKL stimulation. RANKL-induced MSK1 activation at Ser 376 and Thr 581 was also mediated by p38 and ERK. MSK1, as a downstream kinase of p38 and PKA, regulated c-Fos and NFATc1 induction by RANKL through CREB and c-Fos.
In conclusion, we showed that RANKL-induced MSK1 activation at Ser 376 could potentially promote c-Fos and NFATc1 induction upon CREB activation. In this regard, MSK1 could be an intermediator in osteoclastogenesis. Although other studies on site-specific mutagenesis of RANKL-induced activation sites and the role of MSK1 in vivo should be performed, MSK1 can be an important regulator in osteoclast differentiation. This study provides fundamental information for an anti-bone destruction drug design.