
Introduction
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder with progressive and devastating memory impairment. The AD patient brain is characterized by amyloid β protein (Aβ) deposits, neurofibrillary tangles and loss of basal forebrain cholinergic neurons [1]. Both in vivo and in vitro studies have reported the toxic effects of Aβ or Aβ peptide fragments in brain suggesting an important role of Aβ in the pathogenesis of AD [2]. The mechanisms underlying Aβ neurotoxicity are complex but may involve oxidative stresses associated with reactive oxygen species (ROS) generation and decreased levels of glutathione (GSH) and superoxide dismutase (endogenous antioxidants). Aβ has been reported to produce hydrogen peroxide and lipid peroxide in neurons [3], superoxide in microglia [4] and proinflammatory cytokines [5]. This Aβ-induced oxidative stress is indexed by protein oxidation, lipid peroxidation, DNA oxidation, and neuronal cell death [6–8]. Therefore, antioxidants such as α-tocopherol and anti-inflammatory agents such as indomethacin reportedly slow the progression of AD [9, 10].
Aralia elata Seem (Araliaceae) is widely distributed in oriental countries such as Korea, Japan and China. The young shoots of Aralia elata are a popular edible plant in the spring. Several bioactive constituents such as saponins, alkaloids, glycosides, palmitic acid, and linoleic acid from Aralia elata have been reported to have anti-inflammatory and anti-oxidative effects and inhibit neurodegeneration [11–13]. Chaenomeles sinensis (Thouin) Koehne fruit (Rosaceae) is a valuable source of health food and Chinese medicine. Chaenomeles sinensis fruit is rich in dietary fibre, organic acids and bioactive pentacyclic triterpene acids such as oleanolic acid and ursolic acid and large amounts of bioactive phenolic acids and vitamin C [14, 15]. These compounds are related to anti-inflammatory and antioxidant effect of Chaenomeles sinensis fruit [16–18]. Phytochemical researches have revealed that the major constituents of Glycyrrhizae radix (Leguminosae), the root of Glycyrrhiza uralensis, are flavonoids and triterpenoid saponins, which possess diverse pharmacological properties such as anti-ulcer, anti-inflammatory, antispasmodic, antioxidation, anti-cancer, anti-depression and memory enhancement [19–24].
In previous reports, Chaenomeles sinensis fruit was demonstrated to show neuroprotective effects against Aβ (25–35)-induced neurotoxicity in in vivo and in vitro [25]. Glycyrrhizae radix prevented Aβ (25–35)-induced neurotoxicity in cultured neurons [26]. Glycyrrhizae radix have been proved for its memory-enhancing effect and its anticholinesterase activity [23, 27]. Studies have reported neuronal protective effects of Aralia elata in some animal models [13, 28]. Therefore, it was hypothesized that mixture of these three plants, possessing anti-oxidant and anti-inflammatory activities, might show more effective and synergistic neuroprotective effect with the reduction of possible individual toxicity due to large quantity. The present study was conducted to confirm the inhibitory effect of an ethanol extract mixture of Aralia elata, Chaenomeles sinensis fruit, and Glycyrrhizae radix, which was named as ACG, on memory impairment induced by a single intracerebroventricular (i.c.v.) injection of Aβ (25–35) in mouse model of AD-type amnesia [29].
Materials and Methods
The dried and sliced Chaenomeles sinensis fruit and Glycyrrhizae radix were purchased from Daegu Oriental Pharm at Daegu, Korea. The young shoots of Aralia elata were collected at Keryong Mountain in Daejeon, Korea. These plants were identified by Dr. Bangyeon Hwang, Chungbuk National University. Each 200 g of the three plants was mixed, extracted with 95% ethanol (3 L × 24 h × 3) at room temperature and filtered through filter paper (Advantec MFS, CA, USA). The filtrate was concentrated under reduced pressure using a rotary evaporator (Heidolph Instruments GmbH & Co,. Schwabach, Germany) to yield an ethanol extract (ACG, 55 g), which was then stored at room temperature until required. Aβ (25–35) was purchased from Bachem (Bubendorf, Switzerland). Acetylthiocholine iodide and 5,5’-dithiobis-2-nitrobenzoic acid (DTNB) were purchased from Sigma Chemical (St. Louis, MO, USA). Thiobarbituric acid (TBA) was purchased from Tokyo Kasei Kogyo (Tokyo, Japan).
Male ICR mice (5-week-old) were supplied by Daehan BioLink (Chungbuk, Korea) and housed in an environmentally controlled room at 22 ± 2°C and 55 ± 5% RH under a 12-h light/dark cycle with ad libitum access to food and water. Mice were allowed to adapt to the experimental environment for a week.
Memory impairment was induced by i.c.v. injection of 15 nmol of the aggregated form of Aβ (25–35), as previously described [2]. ACG was administered orally 30 min before Aβ (25–35) injection on experimental day 1 (ED1) and then further administered daily for 7 days (to ED8). ACG was suspended in distilled water. Sham-controls were injected i.c.v. with physiological saline instead of 15 nmol of Aβ (25–35). Mice were randomly divided into five groups ; sham-operated (the sham control group), the 15 nmol Aβ (25–35) group, the 15 nmol Aβ (25–35) + 10 mg/kg ACG group, the 15 nmol Aβ (25–35) + 25 mg/kg ACG group, and the 15 nmol Aβ (25–35) + 50 mg/kg ACG groups. Distilled water as vehicle was administered orally to the sham control and Aβ (25–35) control groups using the same schedule as ACG administration.
Passive avoidance test was performed using a two-compartment shuttle chamber device (Avoidance System version 1.1, B. S Technolab, Seoul, Korea), one chamber was illuminated and the other was unlit and equipped with a grid floor and shock generator [2]. Thirty minutes after administering ACG on ED8, the mice were trained to perform the step-through passive avoidance task with the electric shock system activated (0.5 mA, 3 s; the acquisition trial). Retention trials were performed 24 h after the acquisition trial and the step-though latencies were recorded (maximum test time was set at 300 s).
A Morris water maze test was performed to measure spatial learning and memory of the mice according to the Morris method [30] with slight modification. The experimental apparatus consisted of a circular water tank (90-cm diameter, height of 40 cm) filled with water (25 ± 2°C) to a depth of 30 cm. An escape platform with an 8-cm diameter was submerged 1 cm below the surface of the water and placed in the middle of the same quadrant throughout the training phase. Mice were pretested in same condition prior to ACG and/or Aβ (25–35) injection to identify the similar ability to find hidden platform on ED1. Mice were allowed 4 training trials per day after ACG administration for 5 consecutive days (from ED4 to ED8). For each trial, the mice were placed into the water from one of the four starting points on the edge of the tank. The time required to escape onto the hidden platform was recorded. If the mouse located the platform, it was allowed to remain there for 10 s. If the mouse failed to find the platform within 120 s, it would be gently guided to the platform where it was allowed to stay for 10 s, and the escape latency was recorded as 120 s.
After the passive avoidance test retention trial on ED9, mice were deeply anesthetized with diethyl ether, brains were quickly removed, and brain homogenates were prepared using five volumes of 0.1 M phosphate buffer (pH 7.4) in ice bath. Cholinesterase activity and levels of reduced GSH in mouse brain were determined spectrophotometrically using Ellman reagent [31]. Acetylthiocholine iodide was used as a substrate to measure cholinesterase activity. Extent of lipid peroxidation was assayed by measuring TBA reactive substance (TBARS) levels at 532 nm as described by Yoshioka et al. using 1,1,3,3-tetramethoxypropane as a standard [32]. Protein concentrations were determined using Lowry’s method [33].
Results
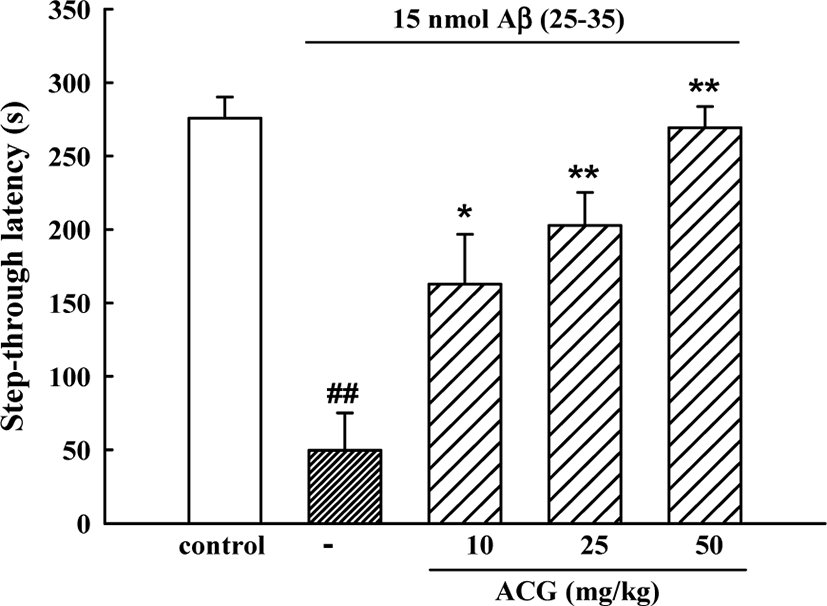
In the initial acquisition trial of the passive avoidance task, the step-through latency did not differ among the 5 groups. However, in the retention trial, step-through latency in the Aβ (25–35)-control group was significantly shorter than that in the sham control group (49.7 ± 25.5 s versus 275.8 ± 14.5 s) indicating that Aβ (25–35) had induced memory impairment. Mice administered ACG daily 8 days showed significant memory improvements (162.7 ± 34.0, 202.7 ± 22.5 and 269.2 ± 14.4 s for 10, 25, and 50 mg/kg, respectively (Fig. 1).
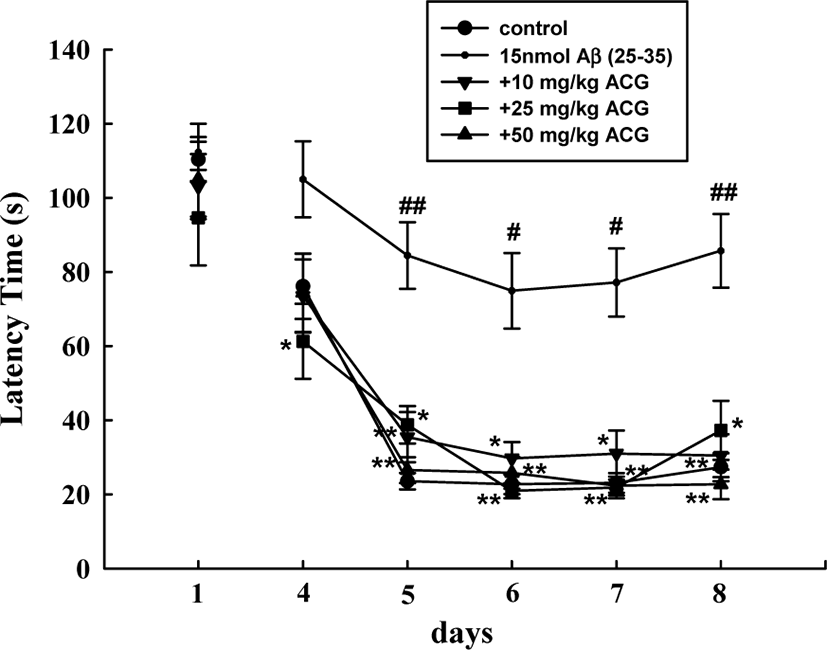
During the first trials using the Morris water maze test, latency time required to reach the hidden platform did not significantly differ among all the groups. The sham mice rapidly learned the location of the hidden platform, reaching it within 30 s on ED5 compared to the first trials (110.3 ± 6.1 s). Mice that received Aβ (25–35) showed a significantly delayed escape latency time from ED5 to ED8 compared to that of the sham-control group, indicating that Aβ (25–35) caused remarkable cognitive deficit. In contrast, chronic administration of ACG (10, 25, and 50 mg/kg) significantly decreased the Aβ (25–35)-induced prolonged latency time (ED5, 6, 7, and 8; Fig. 2).
Cholinesterase activity in the brains of mice in the Aβ (25–35)-treated group was significantly higher than that in the sham control group (9.3 ± 0.5 μmol/h/mg protein versus 5.1 ± 0.3 μmol/h/mg protein). The increased cholinesterase activity was significantly inhibited by 25 and 50 mg/kg ACG, showing 7.5 ± 0.2 and 7.2 ± 0.3 μmol/h/mg protein, respectively (Table 1).
| Group | Dose (mg/kg) | Cholinesterase activity (μmol/h/mg protein) |
|---|---|---|
| Sham | - | 5.1 ± 0.3 |
| Aβ (25–35) | 15 nmol/animal | 9.3 ± 0.5## |
| + ACG | 10 | 8.8 ± 0.2 |
| + ACG | 25 | 7.5 ± 0.2** |
| + ACG | 50 | 7.2 ± 0.3** |
GSH contents of the brains from Aβ (25–35)-treated mice was significantly lower compared to that of the sham group. ACG (25 and 50 mg/kg) inhibited the decrease of GSH levels in Aβ (25–35)-treated brains. TBARS concentration in the brain of Aβ (25–35)-treated mice significantly increased compared to that of the sham group. ACG (25 and 50 mg/kg) suppressed the elevation of TBARS concentration induced by Aβ (25–35) (Table 2).
Discussion
Aβ peptides (1–40 and 1–42 amino acid), cleaved from amyloid protein precursor, are the major constituents of senile plaques occurring in AD and playing a critical role as the principal toxic species to neuronal cell death and breakage of neurite [34]. The 11 amino acid sequence Aβ (25–35) exhibits neurotoxic properties of full-length sequence Aβ (1–42) [35]. It is widely acknowledged that mice i.c.v. injected with Aβ (25–35) can mimic some cognitive deficits in AD [36, 37], showing memory impairment in different behavioral paradigms such as Morris water maze and passive avoidance tests [38, 39]. In the present study, i.c.v. injection of Aβ (25–35) to mice produced memory impairment in the passive avoidance test and Morris water maze test. Chronic treatment with ACG effectively protected the mice against Aβ (25–35)-induced memory deficit.
Cholinergic transmission is crucial to learning and memory, and its alteration is considered main causes of cognitive disorder in AD. I.c.v. injection of Aβ (25–35) in rats resulted in learning and memory deficits that were accompanied by reduction of choline-acetyltransferase activity [40]. Accumulation of Aβ in AD brain is associated with elevation of cholinesterase and results in learning and memory deficits [41]. Thus, some cholinesterase inhibitors, tacrine (Cognex®) and donepezil (Aricept®), have been used for the treatment of AD [41–43]. In the present study, the cholinesterase activity in brain tissue was increased in Aβ (25–35)-treated mice, and ACG (25 and 50 mg/kg) significantly inhibited the increase of cholinesterase. This result suggests that ACG could ameliorate memory impairment by inhibiting cholinesterase activity.
Studies have suggested that oxidative stress is responsible for the onset of the cognitive dysfunction during the progression of AD [3, 4, 7, 44]. Extracellular Aβ accumulation may directly or indirectly alter N-methyl-D-aspartic acid (NMDA) type glutamate receptor-mediated cytosolic increases in Ca2+, which is rapidly taken up by mitochondria and endoplasmic reticulum. Ca2+ overload of mitochondria causes ROS generation [45], and excessive production of ROS and impairment of the ROS removal system in the AD brain result in protein, nucleic acid, and neuronal membrane lipid damage, which in turn, cause neuronal cell death [46]. Therefore, a number of antioxidants have been reported to protect cells from Aβ (25–35)-induced toxicity [9, 47, 48]. GSH, a major intracellular antioxidant, plays a key role in the removal of ROS and protection of cells from oxidative stress. GSH level, thus, is the first indicator for oxidative stress in the brain [49]. TBARS level, commonly used as lipid peroxidation marker, has been elevated in the AD brain tissue [44]. In the present study, brain GSH contents in the Aβ (25–35)-treated group were significantly reduced, and ACG administration significantly prevented these decreases. Additionally, Aβ (25–35)-induced increase in TBARS concentration was significantly decreased by administration of ACG.
The three constituent plants of ACG and their active principles have been reported for neuroprotective activities. It is well known that Aralia elata contains antioxidants, such as congmuyanosides, echinocystic acid and oleanolic acid [12, 50]. Ethyl acetate fraction from Aralia elata (100 mg/kg) improved learning, memory ability, and spatial cognition through inhibition of cholinesterase activity and antioxidant effect [51]. Chaenomeles sinensis fruit is rich in antioxidant components such as flavonoids, phenolics, total triterpenes, and vitamin C [52]. In a previous report, we demonstrated that Chaenomeles sinensis fruit has protective effect on Aβ (25–35)-induced neurotoxicity in in vitro and in vivo [25]. Chaenomeles sinensis fruit inhibited Aβ (25–35)-induced increase of intracellular Ca2+ concentration ([Ca2+]i), ROS generation, and, in result, neuronal death in cultured neurons. Furthermore, Chaenomeles sinensis fruit (50 mg/kg) suppressed Aβ (25–35)-induced increase of brain cholinesterase activity and memory impairment. Glycyrrhizae radix contains many licorice flavonoids including liquiritin, isoliquiritin, liquiritigenin and isoliquiritigenin [53]. Glycyrrhizae radix and isoliquiritigenin prevented Aβ (25–35)-induced neuronal apoptotic death by interfering with the increases of [Ca2+]i and ROS in cultured cortical neurons [26]. Memory enhancing activity of Glycyrrhizae radix in mice was shown at dosage of higher than 150 mg/kg [23, 54]. Maximal effect of ACG was shown at the lower dosage (50 mg/kg) than the sum of maximal effect dosage of each plant. Therefore, it is suggested that the preparation of ACG might reveal synergistic effect of three medicinal plants in protection of Aβ-induced memory impairment through the inhibition of ROS generation and cholinergic dysfunction. Abundant antioxidants contained in this preparation might be responsible for the prevention of Aβ (25–35)-induced neurotoxicity. In conclusion, it is evident that ACG could provide a marked protection against Aβ (25–35)-induced memory deficit. This result may explain the inhibitory action of ACG on the progression of AD.