Introduction
In the early stages of stomach cancer, variations in cellular genes arise as a result of genetic and environmental factors. These variations then lead to abnormal protein expression and eventually, abnormalities in cell physiology, cell division, etc. Stomach cancer causes post-transcriptional modifications, which alter overall functionality not only by genetic variation but also via increased or decreased expression of specific proteins [1]. The incidence of stomach cancer has recently been found to be decreasing gradually. However, it remains the most frequent malignancy in Korea. According to global data, the incidence of stomach cancer is higher in Asia compared with Europe or America. According to 2017 statistics, the incidence of stomach cancer remains high, ranking first in men and fourth in women, highlighting the importance of its early detection and treatment [2]. Progress has been made in methods that can detect stomach cancer by not only early diagnosis, but also simple checks. The reported trend of lowering rates of stomach cancer indicates the role of early detection through endoscopic methods as a result of advancements in medical techniques. This explains the drastic drop in the death rate statistics compared with the incidence rate of stomach cancer [3]. In particular, when stomach cancer is detected at an early stage and is actively treated, the prognosis improves [4].
Genes related to stomach cancer are potential targets for cancer treatment and can serve as important biological markers to determine not only diagnosis or prognosis but also response to treatment. Accordingly, it is necessary to detect candidate genes and study their expression in terms of stomach cancer [1]. Because of the dissemination and proliferation of the next-generation sequencing (NGS) technique, the amount of genomic data is increasing exponentially. As the volume of data is increasing, it is possible that important features that might be missed by general approaches based on statistical analysis are discovered based on machine learning algorithms in artificial intelligence [5, 6]. The application of convolutional neural network (CNN) - a deep learning algorithm having the best performance in image classification of clinical and biological data - can enable the discovery of new biomarkers that can be used for the early diagnosis and prediction of prognosis of stomach cancer [7, 8]. It can also be applied extensively to a variety of basic, clinical, and medical big data [9]. In this study, after discovering candidate genes that influence stomach cancer by principal component analysis (PCA) using RNA-seq data from The Cancer Genome Atlas (TCGA), a public cancer database, we developed a classification model using the CNN algorithm. Using this model, we predicted the diagnosis, vital status, and sample type and estimated the classification accuracy.
The stomach is an important organ for digestion, and mucous membranes on the walls of the stomach protect stomach cells. Stomach cancer typically develops in this mucous membrane (95%), and the stage is determined according to the layer where cancer cells have penetrated. According to TCGA and Asian Cancer Research Group (ACRG) data sets, stomach cancer is categorized into four different types, which are associated with prognosis and have distinct clinical properties. The four genetic types are as follows: genetically unstable type (50%), microsatellite instability with high genetic methylation (22%), genetically stable type (20%), and Epstein–Barr virus type (9%). However, in the case of stomach cancer, relatively little is known about the genetic variation related to the etiopathogenic mechanism compared with other kinds of cancer. Accordingly, few targeted therapies and biomarkers have been developed [10, 11]. Recent studies have shown that microRNAs are related to cancer occurrence in a variety of cancer types. Generally, although messenger RNAs (mRNAs) are composed of thousands of nucleotides, microRNAs consist of 20–22 nucleotides. MicroRNAs and mRNAs function complementarily and control mRNA expression in cells, which is involved in cancer development and progression [12]. Many RNAs do not produce proteins, including microRNAs, some of which control gene expression [13, 14]. Accordingly, tracing variation at the level of gene expression, identifying important gene variation associated with cancer, and constructing prediction models based on genetic variation make it possible to predict the frequency and occurrence of stomach cancer by simple checks [15, 16]. Accordingly, in this study, we combined RNA-seq gene expression data for stomach cancer obtained from TCGA (an open database with clinical data), searched for candidate genes, and analyzed these genes using a deep learning algorithm. In particular, we selected candidate genes that can distinguish between healthy individuals and patients with cancer by PCA, produced a heatmap that represents the expression of candidate genes to construct a model, and applied the CNN classification algorithm. Furthermore, we developed a classification model for survival according to the level of RNA-seq–based gene expression.
Materials and Methods
Data for 334 patients with cancer were obtained from TCGA, a worldwide cancer database. The transcription profiling file, as well as case files, including sample information and clinical information, were used [17]. Next, clinical data, expression data, and case data were combined into a single file based on case ID and file name using Python. The final data set included 334 samples and 60,483 genes.
We used MATLAB as experimental tool to analyze the data obtained from TCGA To extract features, PCA was used. The extracted features were used to generate a heatmap, and learning was performed using the CNN algorithm, a deep learning approach [18–20].
PCA is a widely applied technique for dimensionality reduction, data compression, feature extraction, data visualization, etc. In this study, PCA was used to extract features related to gene expression with major differences among samples. The observed value is a vector x of dimension D, and the data set that is the object of PCA is defined as {xn}, n = 1, 2, · · ·, N. The purpose of PCA is to determine the principal subspace with M < D dimensions, which maximizes the variance of the projected data. The representation of the observed value in this principal subspace becomes a feature vector of observed values. To determine the subspace that satisfies the conditions, we defined the sample mean x̄ and data covariance matrix S as follows.
Using the definitions in (1) and (2), we defined the unit vector u_i on the principal subspace that maximizes the variance of a given data set as follows.
That is, the vector that maximizes the variance of the projected data becomes an eigenvector, ui, of matrix S, and the size of the maximal variance in the direction of the eigenvector becomes the eigenvalue λi. Accordingly, the principal subspace composed of the principal component derived from PCA is composed of an eigenvector of M pieces of maximal eigenvalues for matrix S. In addition, the feature vector value derived from the observed values of xn is given as a coefficient that represents a linear combination with M pieces of the eigenvector for the principal subspace.
CNN is garnering increasing attention in the area of deep learning targeting image data. It has the ability to detect the correlation between adjacent pixels in an image and retain invariance with respect to changes in scaling, such as parallel translation, expansion, and reduction, which are frequently produced in image data. Owing to these strengths, based on CNN images, the approach is extensively applied, e.g., in the recognition of handwriting and the recognition of objects, logos, and features in images. A typical CNN structure is shown in Fig. 1.
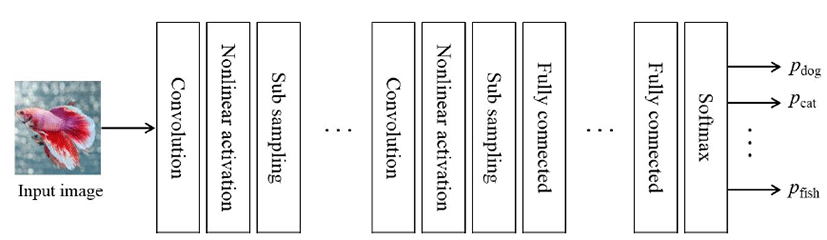
The structure has repeated layers, such as the convolutional layer, which measures the weighted moving sum; the nonlinearity layer, which is composed of an activation function; and a sub-sampling layer, which applies spatial reduction to image data. It also has a neural network layer with a fully connected structure and a Softmax layer, which represents the probability of classification. In the convolutional layer, it performs the convolution calculation, which measures the weighted moving sum for a rectangular area, and the filter size, where the rectangular area proceeds by the stride length. Finally, the result is generated according to the filter number. Next, the result of the convolutional layer becomes the input for the nonlinearity layer activation function; in the case of CNN, it uses the rectified linear unit (ReLU) function, which improves the efficiency of gradient calculation using backpropagation. Subsequently, a sub-sampling is used to reduce the dimension of feature values using max pooling, which chooses a feature value that is the greatest stimulus in the rectangular area with a fixed size. The layers, such as the convolutional layer, nonlinearity layer, and sub-sampling layer, are connected repeatedly and produce a feature vector for the input image. To perform learning using the feature vector, the layers connect the neural network with the fully connected structure. By learning, weighted values that connect each neuron are adjusted to the value that minimizes the sum of square of classification errors. Finally, the Softmax layer represents the classification probability for the input image.
A heatmap is a visual representation of each matrix element using colors for the two-dimensional representation of data matrix values. Here, large values are represented as small rectangles or pixels with a dark color, and small values are represented as bright colors. Because heatmaps are useful for visualization, with a variety of potential color schemes according to the application, they have extensive applications, e.g., for analyses of web page visits, gene expression data, and data visualization. In this study, a heatmap was used to convert the feature vector obtained by PCA into image information needed for CNN.
Results
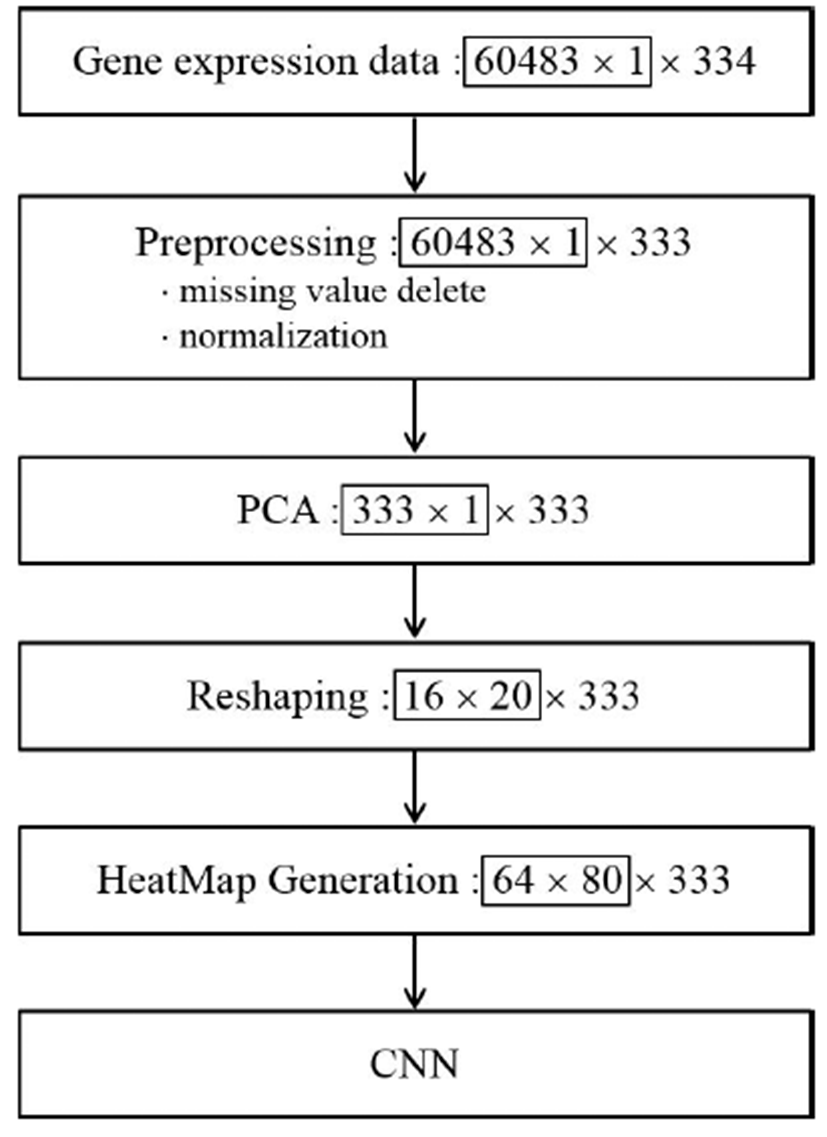
The whole processing procedure for the deep learning system for gene expression data is summarized in Fig. 2 We eliminated noise and singular values and performed normalization for PCA in the preprocessing steps. For the PCA, X conforms to D = 60,483 and N = 334 as a D × N dimensional matrix. Each row of matrix X represents the observed value for one patient, and each column represents the gene expression information for the patient. By PCA, we calculated 333 principal components for each observed value and selected 320 principal components according to the size for subsequent processing. Subsequently, after generating a 16 × 20 matrix, we converted this matrix into a 64 × 80 HeatMap image and input the image into CNN. Fig. 3 shows an example of a HeatMap image for the observed values produced using the HeaTmap function of MATLAB. Fig. 4 shows the structure of CNN for learning.
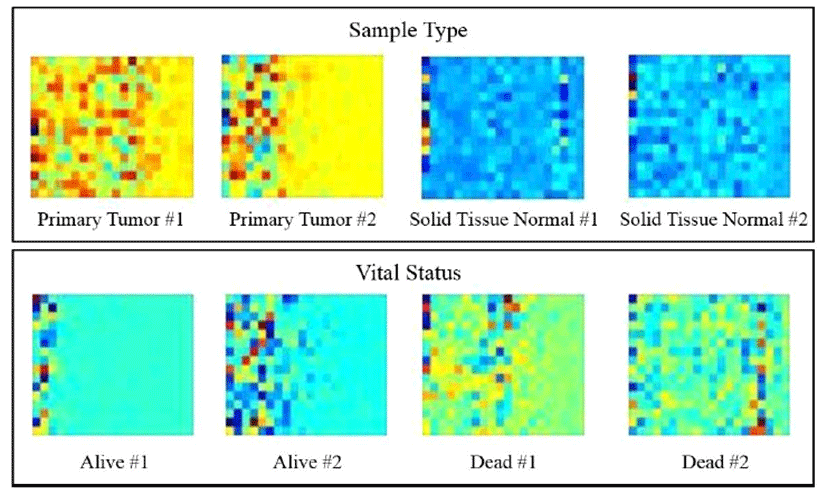
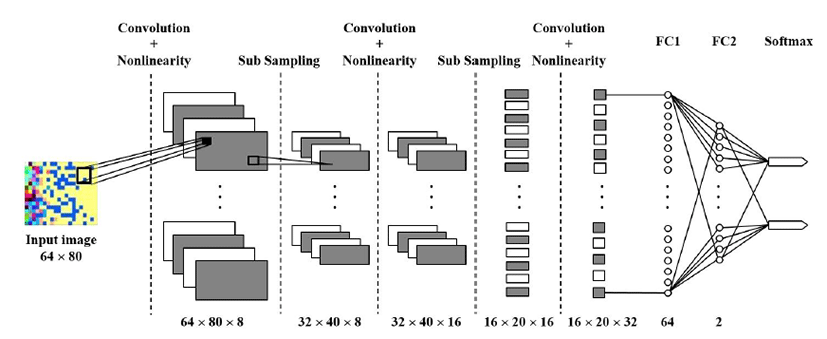
In the case of primary tumor (PT) for sample type, as shown in Fig. 3, the pixels are biased toward the left-hand side with a yellow background. However, in case of solid tissue normal (STN), a line of blue pixels is widely distributed. For live and dead cases (i.e., the vital status category), a similar pattern was detected. These results indicated that principal components for gene expression extracted from PCA differed with respect to image and color among categories. Next, Fig. 4 shows the CNN structure for different learning steps.
Layers such as convolutional, nonlinear activation (ReLU), and subsampling (Max pooling), are repeated thrice after passing through two fully connected layers, and Softmax values are outputs. Moreover, to avoid overfitting in the neural network, dropout with a probability of 0.5 was inserted between fully connected layer 1 (FC1) and fully connected layer 2 (FC2).
The detailed structure of the CNN and hyperparameter values that influence learning at each stage are as follows.
Convolutional layer: Filter size (3), Filter number (8), Padding = 1 ReLU + Maxpooling: size (2), Stride = 2
Convolutional layer: Filter size (3), Filter number (16), Padding = 1 ReLU + Maxpooling: size (2), Stride = 2
Convolutional layer: Filter size (3), Filter number (32), Padding = 1
FC1: Output (64)
FC2: Output (2)
We used 234 randomly selected data points (70% of the whole data set) for learning, and the learning options were set to 40 for Epoch and 32 for MiniBatchSize.
Recognition result about 333 of data except one missing value among 334 of data is shown in Table. 1.
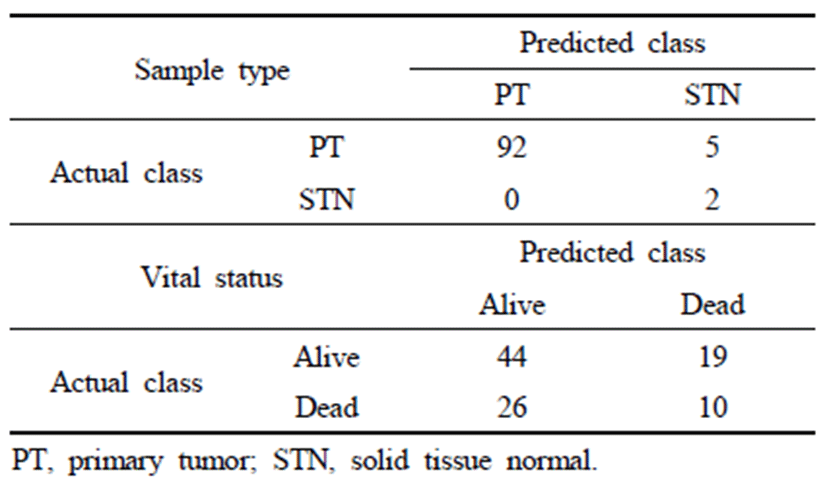
Table 1 shows the verification results for 99 datapoints, i.e., 30% of the whole data set. For distinguishing between the sample types PT and STN, the accuracy was 95.96%, and the accuracy was greater for PT than for STN. With respect to vital status (i.e., alive and dead), the accuracy was 50.51%, which was lower than the accuracy for sample type. Accordingly, we found that the differences in gene expression with respect to sample type are distinct from those for vital status. In both cases, the accuracy of the validation data is lower than the accuracy for the learning data. This indicates that overfitting resulted from the small sample size (N = 333) and high dimensionality (D = 60,483) of the gene expression data. Accordingly, it is possible to resolve the overfitting issue and improve the generality of the method by obtaining gene expression data from more patients.
Discussion
In this study, we performed feature classification of stomach cancer using gene expression data of TCGA. First, applying the PCA method, we selected candidate genes that are principal components explaining gene expression data of stomach cancer. We converted selected candidate gene expression information corresponding to principal components into heatmap images and measured the accuracy of prediction of representative prognosis of stomach cancer patients using the CNN algorithm in deep learning. A classification was obtained by PCA, and the prediction accuracy using deep learning was lower than the accuracy of recognition problems, such as number recognition. This can be explained by the wide variety of causes of stomach cancer. The disease may be too complex to explain using simple gene expression variation. Studies on stomach cancer biomarkers have been conducted, including synthetic literature reviews, leading to the discovery and classification of causal variants directly relevant to stomach cancer or rare variants with low frequencies. In the future, based on these results, analyses of the functions of candidate genes and correlations in expression based on deep learning and big data may facilitate the early detection of stomach cancer and the development of treatment approaches and methods to predict prognosis.