Introduction
Alzheimer’s disease (AD) is characterized by neuronal loss and extracellular senile plaques, which primarily consist of β-amyloid protein (Aβ), a 39-43 amino acid peptide derived from amyloid precursor protein [1]. The deposition of Aβ in the pathogenesis of AD is invariably associated with oxidative stress and inflammatory responses [2]. Antioxidants such as α-tocopherol protect against Aβ-induced cytotoxicity as well as against development of learning and memory deficits [3]. Additionally, anti-inflammatory agents such as indomethacin have been reported to slow the progression of AD [4]. Furthermore, injection of Aβ (25-35) into the cerebral ventricle of mice was found to result in learning and memory deficits that were accompanied by decreased choline acetyltransferase and increased cholinesterase activity, suggesting that accumulation of Aβ disrupts cholinergic activity and causes the cognitive impairments associated with AD [5].
Actinidia arguta (sieb. Et Zucc.) Panch (Actinidiaceae) is a smooth-skinned grape-sized kiwifruit native to Korea, northern China, Siberia and Japan. The fruits, leaves, stems and bark of A. arguta have traditionally been used for the treatment of inflammatory diseases and gastrointestinal diseases in Korea [6]. Moreover, A. arguta have been reported to possess anti-oxidant, antiapoptotic, antiinflammatory and anti-allergic properties [7-10], as well as to contain various anti-oxidants including catechins, vitamin C, carotenoids, chlorophyll, anthocyanin, and other polyphenols [11-14]. Antioxidative products are commonly regarded as potential neuroprotective agents because they improve a number of pathological processes, including ROS formation and inflammation [15]. Therefore, this study was conducted to investigate the neuroprotective effects of the leaves and stems of A. arguta in vitro using cultured neurons and in vivo in experimental animals.
Materials and Methods
The leaves and stems of A. arguta were collected from Keryong Mountain in Daejeon, Korea and identified by Professor KiHwan Bae of the College of Pharmacy, Chungnam National University, Korea. Dried leaves and stems of A. arguta (4 kg) were extracted three times with ethanol at room temperature for three days, filtered, and concentrated to yield an ethanol extract (300 g; yield: 7.5%), which was stored at room temperature until required. Aβ (25-35) was purchased from Bachem (Bubendorf, Switzerland). 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT), Dulbecco's modified Eagle's medium (DMEM), Joklik-modified Eagle's medium, hydrogen peroxide (H2O2) and poly-L-lysine were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Hoechst 33342 dye was purchased from Molecular Probes Inc. (Eugene, OR, USA). Fetal bovine serum was purchased from JRS Biosciences (Lenexa, KS, USA).
Experimental animals
Pregnant Sprague-Dawley (SD) rats and male ICR mice were purchased from Daehan BioLink Co. Ltd. (Chungbuk, Korea) and housed individually and in groups of 10, respectively, in environmentally controlled rooms at 22°C ± 2°C under a relative humidity of 55 ± 5% and a 12-h light/dark cycle while provided with food and water ad libitum. The procedures involving experimental animals complied with the animal care guidelines of the United States National Institutes of Health and the Animal Ethics Committee of Chungbuk National University.
Primary cortical neuron cultures were prepared using embryonic day 15 to 16 SD rat fetuses [16]. Neurotoxicity experiments were conducted on neurons that had been grown for 4–6 days in culture. An Aβ (25-35) stock solution of 2 mM was prepared in sterile distilled water, stored at -20°C, and incubated for more than 2 days at 37°C to aggregate before use. Cultured neurons were treated with 10 μM Aβ (25-35) in serum-free DMEM at 37°C for 24 h to produce neurotoxicity. A. arguta (1, 10 and 50 μg/ml) was applied 20 min prior to treatment with 10 μM Aβ (25-35) and was also present in the medium during Aβ (25-35) incubation. To measure the H2O2-induced neuronal death, cultured neurons were treated with 100 μM H2O2 for 15 min in a HEPES-buffered solution containing HEPES (8.6 mM), NaCl (154 mM), KCl (5.6 mM), and CaCl2 (2.3 mM) at pH 7.4. After exposure to H2O2, the neurons were washed and further incubated in H2O2-free and serum-free DMEM for 12 h (post-incubation). A. arguta (1, 10 and 50 μg/ml) was added 20 min prior to treatment with H2O2 and was also present in the buffer during the H2O2 incubation and post-incubation periods. A. arguta was dissolved in DMSO to a concentration of 100 mg/ml, then further diluted in experimental buffers. The final concentration of DMSO was less than 0.1%, which did not affect cell viability. At the end of the incubation period, an MTT assay and Hoechst 33342 staining were performed to measure neuronal cell death and apoptosis, respectively, as previously described [16].
Intracerebroventricular (i.c.v.) injection of the aggregated Aβ (25-35) (15 nmol) was performed to induce memory impairment in mice, as previously described [17]. A. arguta (10, 25 and 50 mg/kg) suspended in distilled water was orally administered to 5-week-old ICR mice 30 min before Aβ (25-35) injection, then further administered once daily for 13 days. A passive avoidance apparatus (Avoidance System Version 1.1, B. S. Technolab Inc, Seoul, Korea) was used to measure memory acquisition as previously described [17]. Mice were trained on a step-through passive avoidance task 30 min after administration of A. arguta on day 13 of i.c.v. injection of Aβ (25-35). A retention trial was given 24 h after the acquisition trial.
Upon completion of the retention trial of the passive avoidance test, mice were anesthetized with diethyl ether and their brains were quickly removed. Brain homogenates were then freshly prepared in an ice bath with a 15- fold volume of a 0.1 M phosphate buffer containing 1% triton (pH 8.0). Cholinesterase activity in the brain was spectrophotometrically determined by the Ellman method [18] as modified by Padilla et al. [19]. Briefly, 5 μl of homogenate and 300 μl sodium phosphate buffer (0.1 M, pH 8.0) containing 0.3 mM DTNB (final concentration in assay 0.25 mM) were mixed, allowed to stand for 5 min, and then amended with 50 μl substrate solution (8.45 mM acetylthiocholine iodide; final 1.2 mM). The increase in the rate of absorbance was monitored for 5 min at 410 nm at 25°C in a microplate reader (TECAN Sunrise, GMBH, Austria). Cholinesterase activity was calculated using an extinction coefficient of 13.6 mM-1cm-1 for 5-thio-2-nitrobenzoic acid, and was expressed as μmol/h/mg protein after protein assay by Lowry's method [20].
Results
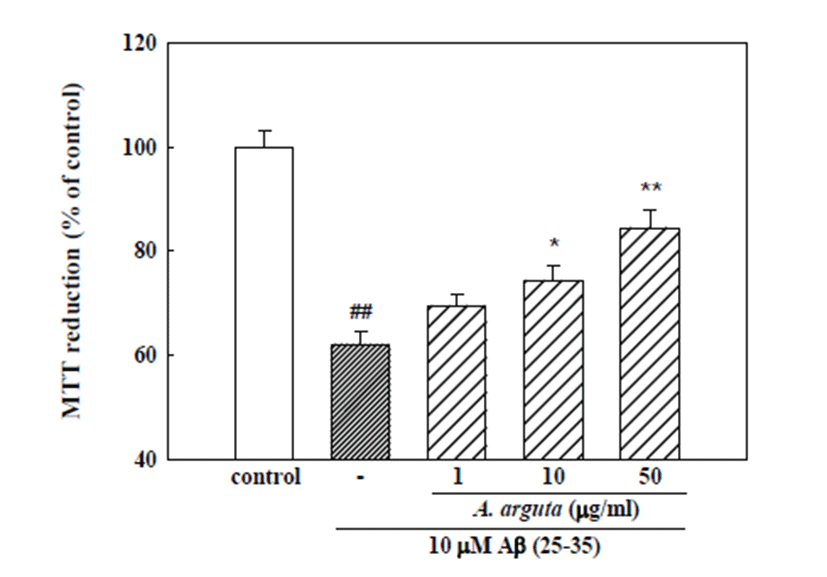
Cultured cortical neurons exposed to 10 μM Aβ (25-35) for 24 h showed 62.0 ± 2.5% absorbance relative to the untreated controls in the MTT assay (Fig. 1), indicating that Aβ (25-35) caused neuronal cell death. Pretreatment of cortical neurons with 10 and 50 μg/ml A. arguta reduced the neuronal death induced by Aβ (25-35) (absorbance, 74.2 ± 3.1% and 84.4 ± 3.5% of control, respectively; Fig. 1).
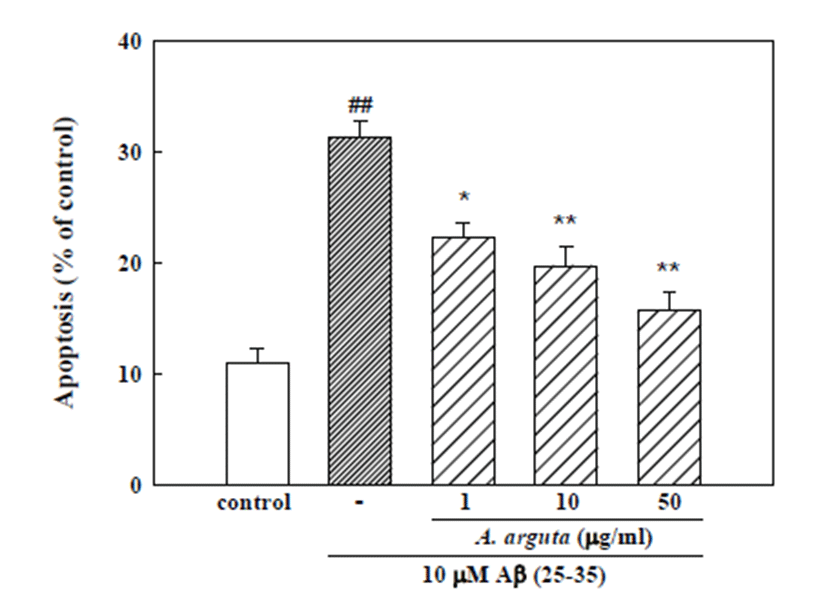
An additional experiment was performed with Hoechst 33342 staining to detect condensed or fragmented DNA, which is indicative of Aβ (25-35)-induced neuronal apoptotic death. Treatment of neurons with 10 μM Aβ (25-35) induced apoptosis of 31.2 ± 1.4% of the total population of cultured cortical neurons, while only 11.0 ± 1.3% of the neurons in control cultures were aptotic. The addition of A. arguta (1, 10 and 50 μg/ml) significantly decreased the Aβ (25-35)-induced apoptotic cell death to 22.2 ± 1.4, 19.7 ± 1.8 and 15.7 ± 1.7%, respectively (Fig. 2).
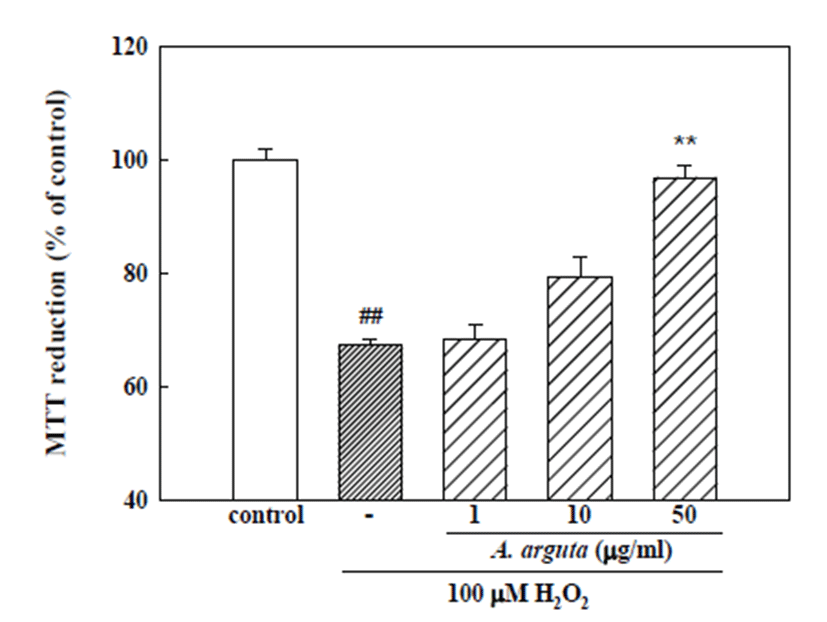
Cultured cortical neurons exposed to 100 μM H2O2 for 15 min showed 67.3 ± 1.0% absorbance relative to the untreated controls in the MTT assay (Fig. 3), indicating that H2O2 caused neuronal cell death. Pretreatment of cortical neurons with A. arguta (50 μg/ml) reduced the neuronal death induced by H2O2 (absorbance, 98.2 ± 2.3% of control; Fig. 3).
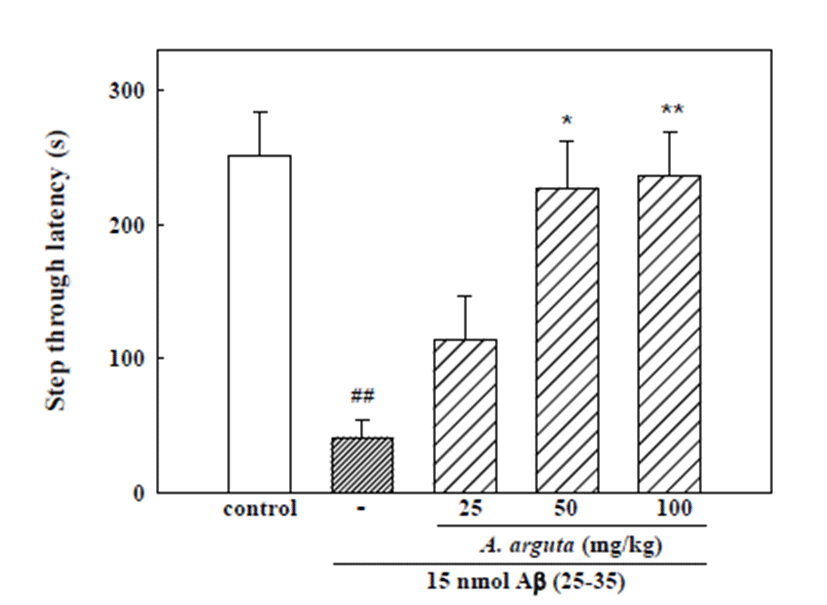
In the acquisition trial of the passive avoidance task, step-through latency did not differ among the five groups (control, 15 nmol Aβ (25-35), 15 nmol Aβ (25-35) + 25 mg/kg A. arguta, 15 nmol Aβ (25-35) + 50 mg/kg A. arguta, 15 nmol Aβ (25-35) + 100 mg/kg A. arguta; data not shown). The step-through latency of the Aβ(25-35)- treated group in the retention trial significantly decreased to 41.2 ± 13.2 s, while it was 251.5 ± 31.7 s in the control group, indicating that Aβ (25-35) induced memory impairment in mice. Chronically administered A. arguta markedly protected against the memory impairment induced by Aβ (25-35). The step-through latency in groups administered A. arguta at doses of 25, 50 and 100 mg/kg was 114.0 ± 32.8, 226.8 ± 34.4 and 235.8 ± 32.2 s, respectively (Fig. 4). Cholinesterase activity in brains exposed to 15 nmol Aβ (25-35) increased significantly; however, treatment with 100 mg/kg A. arguta led to a significant decrease in cholinesterase activity compared with the Aβ (25-35) group (Table 1). To determine if A. arguta treatment affects general motor functions, we measured the spontaneous locomotor activity of the mice. Neither A. arguta nor Aβ (25-35) significantly influenced the locomotor activity (data not shown), indicating that the ob- served memory improvement was not due to immobility that might be caused by A. arguta administration.
Discussion
Alzheimer’s disease is an irreversible neurodegenerative disorder characterized clinically by the loss of cognitive function and pathologically by the appearance of senile plaques and neurofibrillary tangles [21, 22]. Aβ peptides 1-40 and 1-42 amino acid, which are cleaved from amyloid protein precursor (APP), are the major constituents of senile plaques occurring in AD and play critical roles as the principal toxic species responsible for neuronal cell death and neurite breakage [23]. Many studies have reported that Aβ protein acts as a potent neurotoxin both in vitro and in vivo [24]. Therefore, Aβ (25- 35) was used to induce neuronal cell death and assess the protective effects of A. arguta against neuronal cell death and memory impairment in the present study.
The results presented herein provide evidences that Aβ (25-35)-induced apoptotic neuronal death was prevented by the leaves and stems of A. arguta. Specifically, MTT assay revealed that ethanol extract of the leaves and stems of A. arguta significantly inhibited Aβ (25-35)-induced neuronal cell death in cultured cortical neurons. A. arguta also significantly decreased the Aβ (25-35)-induced apoptosis of cultured neurons measured by Hoechst 33342 staining. Aβ-induced neurotoxicity has been attributed to Ca2+ influx followed by generation of reactive oxygen species (ROS) such as O2- and H2O2, which was blocked by treatment with Ca2+ antagonist and ROS scavenger [25, 26]. These studies suggest that the deposition of Aβ in the pathogenesis of AD is invariably associated with oxidative stress. Therefore, the preventive effects of A. arguta against H2O2-induced cultured neuronal cell death were examined in the current study. The results revealed that A. arguta reduced H2O2-induced neuronal cell death, suggesting that the neuroprotective effects of A. arguta may be attributable to its antioxidant activity.
Aβ (25-35) preferably induces impairments of spatial and non-spatial short-term memory, and these effects remain evident for up to 6 months after even a single i.c.v. injection of the peptide [27]. This model has been used to investigate pathogenesis and therapeutics of AD. Memory impairment in the passive avoidance test was also confirmed in mice two weeks after the i.c.v. injec- tion of Aβ (25-35) in the current study. Chronic treatment with A. arguta (50 and 100 mg/kg) effectively protected the mice against Aβ (25-35)-induced memory deficit. This result is consistent with its protective effect on Aβ (25-35)-induced neurotoxicity in cultured neurons. Studies have indicated that oxidative stress in the pathology of AD is responsible for the onset of cognitive dysfunction as well as progression of the disease [28, 29]. A high level of Aβ is responsible for the increased appearance of ROS such as superoxide (O2-) and NO in AD, which produce ONOO- via a rapid interaction [30, 31]. A scavenger of ONOO- was confirmed to protect against Aβ (25-35)-induced memory impairment [32]. A. arguta inhibited H2O2-induced neuronal cell death in the present study. A variety of anti-oxidant compounds such as catechin and epicatechin have been isolated from A. arguta [14, 33]. These compounds have been confirmed to exert significant neuroprotective activities against Aβ- and H2O2-induced neurotoxicity in cultured neurons [34, 35]. Furthermore, A. arguta was shown to protect cultured cortical neurons against glutamate-induced neurotoxicity via inhibition of [Ca2+]i increase and ROS generation in our previous study [36]. Therefore, the neuroprotective effects of A. arguta on Aβ (25–35)-induced neuronal cell death and memory impairment could be attributable to these antioxidant components in the current study.
Cholinergic transmission is crucial to learning and memory, and its alteration is considered one of the main causes of cognitive disorders such as AD [5]. Aβ accumulation associated with cognitive impairment in AD is accompanied by an increase in cholinesterase activity [37]. Cholinesterase activity in the brains of Aβ (25-35)-injected mice significantly increased, but was inhibited by chronic administration of A. arguta in the present study. These results suggest that A. arguta could ameliorate memory impairment by inhibiting cholinesterase activity. The results of the present study provide a mechanistic explanation for the protective effects of A. arguta against Aβ (25-35)-induced neuronal cell death and memory impairment. In conclusion, these results demonstrate the possibility of A. arguta having neuroprotective effects in AD brains that prevent progression of the disease and may provide a pharmacological basis for its therapeutic use in the prevention of neurodegeneration in AD.