INTRODUCTION
Nephroblastoma is believed to originate from the metanephric blastema in the renal cortex [1]. In mammals, nephrons and renal interstitial connective tissue are of mesodermal origin, and the metanephric blastema has the potential to differentiate into either nephron epithelium or interstitial tissue [2].
Although nephroblastoma, also known as Wilms’ tumor, is a common pediatric renal neoplasm in humans, it rarely occurs spontaneously in young rats [3]. In rats, nephroblastoma is typically associated with chemical carcinogens, such as N-ethyl-N-nitrosourea and N-methyl-N-nitrosourea, particularly in immature rats [2, 4, 5]. Nephroblastoma is considered malignant due to the presence of deeply basophilic blastema cells with partial differentiation into tubules, although metastases are infrequent [4–6].
Histopathologically, nephroblastomas typically display a characteristic triphasic pattern, comprising epithelial, stromal, and blastemal components [7]. The tumor is composed of densely packed blastema-like basophilic epithelial cells. These pleomorphic cells have scant cytoplasm and often form an island, with tubular structure arranged at their center within the interconnecting stromal meshwork of mesenchymal tissues [1, 7]. Primitive blastema elements, such as central tubules lined by a single layer of cuboidal to columnar cells and primitive glomerular structures, may be present [1]. Larger neoplasms are typically well-vascularized and may display areas of necrosis and hemorrhage, displacing much of the renal parenchyma [1, 7]. In comparison, Wilm’s tumor in juvenile humans contains both neoplastic epithelial and mesenchymal elements, whereas nephroblastoma in young rats is surrounded by non-neoplastic connective tissue [1].
Since mesenchymal tumors often contain epithelial components and resemble nephroblastoma, the identification of mesenchymal cell proliferation within preexisting nephrons or epithelial tissue is helpful in examining the tumor origin and the features of proliferating cells and differentiating this tumor from other renal tumors, especially renal mesenchymal tumors composed of neoplastic mesenchymal tissues [1, 8].
Immunohistochemistry of cytokeratin, Wilms Tumor 1 (WT1), and Ki-67 can be used for tubular epithelium, blastemal cells, proliferating cell marker, respectively, and α-smooth muscle actin (α-SMA) and vimentin can aid the differential diagnoses [9, 10].
Herein, we present a case of nephroblastoma occurred at the renal papilla of a male rat.
CASE REPORT
A papillary mass was found in the right kidney of a male 10-week-old Sprague-Dawley rat. This case was found at a control group of a repeated-dose administration study to assess the potential toxicity of a substance. Food and tap water were provided ad libitum. The control animals received dimethyl sulfoxide (DMSO) as a vehicle.
Gross examination of the kidney revealed a neoplastic papillary mass (7.37 × 3.27 × 6.19 mm). No other remarkable findings were detected in the body weights, food/water consumption, clinical signs, organ weights, and clinical pathology, including complete blood chemistry, coagulation profile, and blood chemistry. For the histopathological examination, the kidney, including the mass, was fixed in 10% phosphate-buffered formalin, routinely processed for paraffin embedment, and sectioned for hematoxylin and eosin staining. For immunohistochemistry, the paraffin sections were dewaxed and heated at 98℃ in Target retrieval solution (Dako target retrieval solution, S236984-2, Agilent, Santa Clara, CA, USA) for 5 min using a microwave oven. After treatment with 5% skim milk in phosphate-buffered saline for 50 min, primary antibodies were applied. The sections were incubated with primary antibodies overnight at 4℃ and treated with the secondary antibody (Dako REAL link-biotinylated secondary Ab2, K5001, Agilent). Labeling was visualized with 3,3’-diaminobenzidine. The antibodies against Ki-67 as a tubular epithelium and proliferating cell marker, α-SMA as a vascular smooth muscle and epithelial-mesenchymal transition marker, and vimentin as a mesenchymal cell marker were used (Table 1).
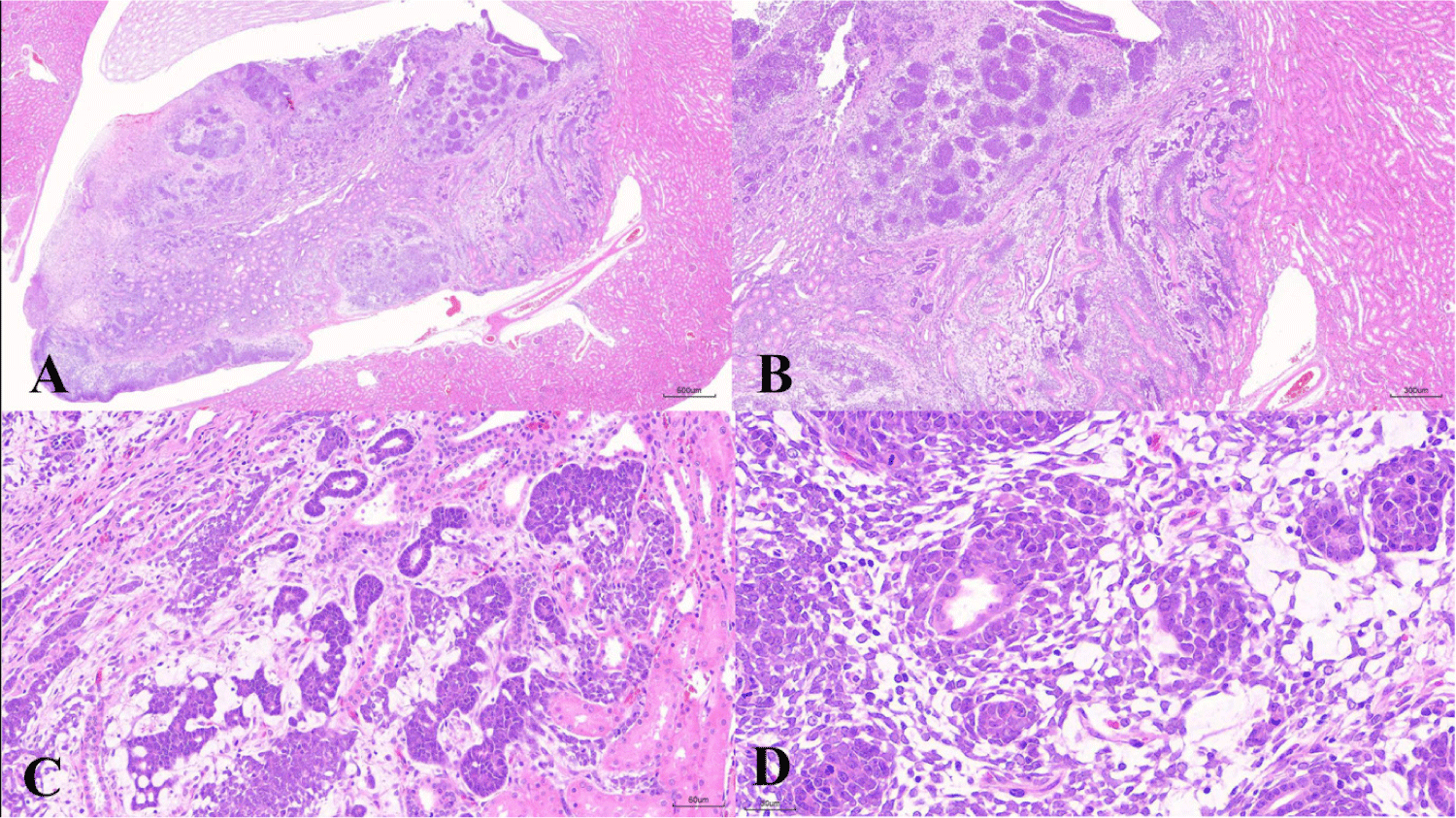
Microscopically, a neoplastic papillary mass protruding from the outer medulla into the pelvis was observed (Fig. 1A), and the neoplastic lesion comprised epithelial and blastema elements (Fig. 1B). In epithelial elements, basophilic neoplastic cells show a high nucleus-to-cytoplasm ratio and tubular growth patterns with small elongated or convoluted tubules. The surrounding interstitial tissue appeared loose and myxomatous (Fig. 1C). In blastemal elements, blastemal cells are often arranged in aggregates or nests with closely packed basophilic polygonal-to-spindloid primitive cells. A primitive glomerulus, well-differentiated eosinophilic tubules located at the center of the blastema elements, and mitotic figures were observed (Fig. 1D).
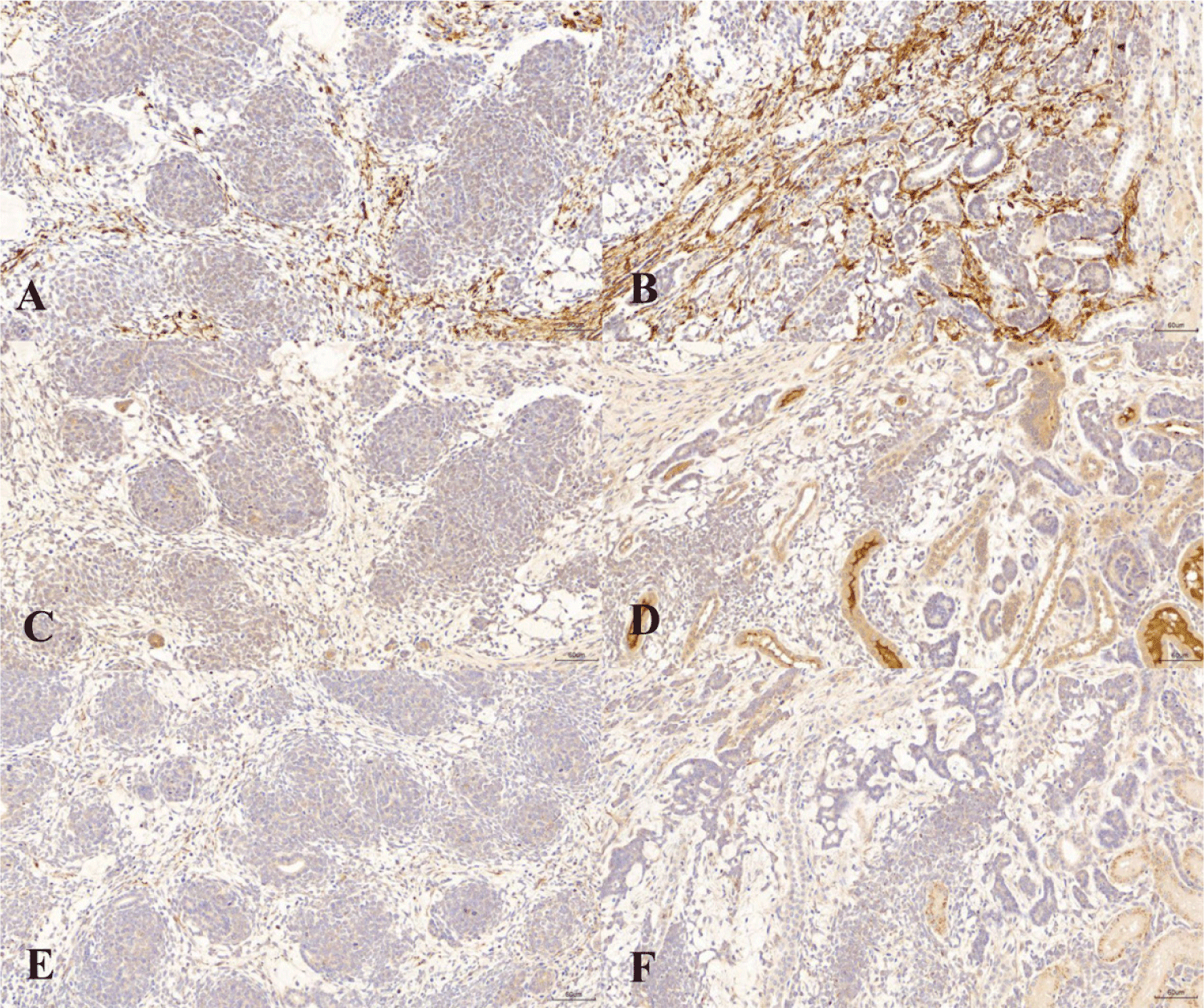
Immunohistochemical staining showed that Ki-67 was strongly expressed in both the blastemal and epithelial elements (Fig. 2A and B). The staining index of Ki-67 was 16.2, which was the average number of positive cells per 100 cells in 10 random fields. α-SMA and vimentin stainings were negative in both blastemal elements and epithelial elements (Fig. 2C, D, E, and F).
DISCUSSION
Nephroblastomas originate from metanephric blastemal elements in the renal cortex and seldom arises from or remains localized to the renal pelvis [6]. While tumors such as carcinosarcoma are infrequently observed in the rat renal pelvis, cases of nephroblastoma specifically confined to this area are exceptionally rare [1, 3, 7].
Nephroblastomas are typically associated with pediatric renal tumors in various animals, including pigs, chickens, dogs, cats and cattle [11]. However, its manifestation in laboratory rats, particularly in preclinical studies, is rare, with an incidence of approximately 0.1% in Sprague-Dawley rats [5, 12]. In domestic animals, this tumor generally has a poor prognosis with metastasis often involving the lymphatic system, lungs, and liver [13]. Although this tumor is rare in rats and the prognosis in rats is less frequently reported in rats, its metastatic tendency and typical late detection following clinical signs onset suggest a similarly poor prognosis [1, 11].
Despite its rarity in rats, nephroblastoma poses a unique challenge in toxicology assessments because of its potential impact on study outcomes and its tendency to remain undetected until clinical signs emerge later in life, complicating interpretations of the tumor as either a spontaneous or treatment-related lesion when observed only in the high-dose group [11, 12].
DMSO is regarded by the international agency for research on cancer (IARC) as non-carcinogenic to humans and is not included on any carcinogen lists of IARC [14]. Additionally, the Salmonella/microsome test results indicate no carcinogenic or mutagenic potential for DMSO, and it is commonly used as a neutral solvent in Ames mutagenicity tests [14, 15]. Moreover, the literatures linking chronic DMSO administration to the development of nephroblastoma, especially in young animals, are extremely limited [13, 16]. Consequently, in this case, it is not considered that the DMSO played a role as a carcinogen or caused nephroblastoma in this case.
Differential diagnosis is crucial to rule out other renal tumors, especially originating from renal pelvis, including renal mesenchymal tumors, renal carcinoma, transitional cell tumors, squamous cell carcinoma, carcinosarcoma, rhabdomyosarcoma, osteosarcoma, and chondrosarcoma [3, 7, 11]. Negative expressions of α-SMA and vimentin support the exclusion of mesenchymal tumors such as renal mesenchymal tumors [17]. In addition, the proliferation of blastemal elements supports the exclusion of epithelial tumors including transitional cell tumors, renal carcinoma and carcinosarcoma [3]. Additional immunohistochemistry using antibodies against WT1 and cytokeratin is required to gain additional information about the characteristics of this tumor and elucidate the underlying mechanisms of nephroblastoma development [8–10].
Based on the observed histopathological features and immunohistochemistry results, we diagnosed the tumor as renal nephroblastoma. In the present case, the tumor was observed in the renal papilla rather than in the renal cortex. As nephroblastomas are known to originate from metanephric blastema cells in the renal cortex, the reason why the main body of the tumor occurs in the renal papilla instead of the cortex should be further elucidated.