INTRODUCTION
Drug-induced liver injury (DILI) is a leading cause of preclinical or clinical drug wastage and acute liver failure [1]. Nearly 100 marketed drugs can potentially induce various types of liver injury, ranging from cell necrosis to fibrosis, or a combination thereof [2]. The severity of DILI can fluctuate based on factors such as the patient’s age, gender, genetics, and underlying medical conditions. Unfortunately, the demands by regulatory agencies such as the Food and Drug Administration (FDA) for live animal testing during preclinical drug development can only detect less than half of human DILIs mainly due to species-specific differences in drug pathways and an impossibility to accurately replicate human genetics and disease conditions [3]. Challenges intrinsic to animal-based drug screening have underscored the increasing importance of in vitro human liver cultures over the past decade or so, with in vitro human liver culture becoming a cornerstone of drug discovery [4]. The process of drug discovery has historically relied on both in vitro and in vivo methods to assess the safety of compounds, with recent emphasis on high-throughput and cost-effective approaches such as cell-based screening assays. Traditionally, these in vitro cell-based assays have been performed using 2D monolayer cell culture, which are a poor mimic of in vivo conditions. Many biological functions, such as cell-cell and cell-substrate contact, are lacking in 2D monolayer cultures. This changes cell signaling pathways and reduces or deficiency of enzymes involved in metabolizing xenobiotics [5., 6]. By imitating the in vivo organization of natural tissues and organs, it is increasingly recognized that cells culture in 3D environments more closely mimic normal cell fuction [7.-9]. In addition, many studies have shown that hepatocytes cultured in 3D form have similar structure and function to liver [10]. As a result, a great deal of effort has gone into the development of a variety of 3D cell models that hold great promise for applications in drug discovery, stem cell research, safety studies and many other cell-based analyses, bridging the gap between traditional 2D monolayer cell culture models and animal models. HepG2 cells remain popular due to their ease of handling and widespread use as one of the oldest and most well-known hepatic cell lines [11]. Previous studies have shown that culturing HepG2 cells for at least 72 hours is sufficient metabolic capacity for in vitro toxicity studies [12., 13]. However, HepG2 cells are known to have low activities of several metabolic enzymes and low expression levels of genes encoding cytochrome P450 enzymes (CYPs) that are essential in drug metabolism [12]. On the other hand, HepaRG cells derived from human hepatoma cell express functional liver markers including various CYPs and phase II enzymes and nuclear receptors. Additionally, HepaRG cells have been shown to be effective surrogates for primary human hepatocytes (PHHs), particularly in CYP3A4 induction studies [14]. In this study, we cultured HepG2 and HepaRG cells and exposed them to hepatotoxicants amiodarone HCl (ADR) and acetaminophen (AAP) to compare their responses. Moreover, we cultured cells in both formats of 2D and 3D spheroids to compare cytotoxicity and hepatic characteristics following drug treatment, aiming to identify which cells and culture types would be the most suitable for drug screening studies.
MATERIALS AND METHODS
The ADR and AAP were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hepatotoxicants were chosen based on published information on hepatotoxic potential and mechanism of action [15.-17]. CYP inducers Omeprazole and Rifampin were purchased from Sigma-Aldrich [18]. A concentrated stock solution of dimethylsulfoxide (DMSO, Sigma-Aldrich) was used to dissolve all chemicals and diluted with cell culture medium. Medium containing a final concentration of 0.1% DMSO was used for the untreated control group.
The human hepatocellular carcinoma cell line (HepG2) was purchased from ATCC (Manassas, VA, USA). HepG2 cells were cultured in Eagle’s minimum essential medium (EMEM, ATCC) supplemented with 10% fetal bovine serum (FBS, Merk Millipore, Burlington, MA, USA) and 1% penicillin and streptomycin (Merk Millipore). HepG2 cells were cultured in T-75 cell culture flask (Corning, New York, NY, USA) at 37°C in a 5% CO2 humidified incubator. Cryopreserved HepaRG cells (Gibco, Braunschweig, Germany) were purchased from Thermo Fisher Scientific and maintained according to the manufacturer’s instructions. HepaRG cells were cultured in William’s E medium (Gibco, Billings, MT, USA) added with HepaRG™ Thaw, Plate & General Purpose Medium supplement (Gibco) and 1% penicillin and streptomycin (Merk Millipore). HepaRG cells were cultured in T-75 cell culture flasks (Corning) at 37°C in a 5% CO2 humidified incubator. When HepG2 and HepaRG cells reached about 80% confluency, cells were trypsinized (0.25% Trypsin-EDTA, Gibco) for 5 min, added with medium to neutralize trypsin, and centrifuged at 218 × g for 5 min at 4°C. The supernatant was removed and cells were re-suspended in medium. These cells were seeded at a density of 1.5 × 104 cells/well in a total volume of 100 μL in ultra-low attachment 96-well plates (BD Bioscience, Franklin Lakes, NJ, USA) and incubated at 37°C in a 5% CO2 humidified incubator. For CYP enzyme induction, cells were cultured in ultra-low attachment 96-well plates. Omeprazole was used as a CYP1A2 inducer. Rifampicin was used as an inducer for CYP2C9 and CYP3A4. When cell density reached more than 80%, cells were incubated according to the condition listed in Table 1 to induce the expression of CYP enzymes.
| CYP enzyme | Inducer | Final concentration (μM) | Incubation time |
|---|---|---|---|
| CYP1A2 | Omeprazole | 0, 25, 50, 100 | |
| CYP2C9 CYP3A4 | Rifampin | 0, 10, 25, 50 | 48 hr |
CellTiter-Glo® 2D Cell Viability Assay and CellTiter-Glo® 3D Cell Viability Assay (Promega, Madison, WI, USA) were used for the measurement of ATP as an indicator of viability, producing a luminescent output which was reported to be more sensitive than other methods [19., 20]. These assays lyse cells using proprietary lytic component. Promega has shown that the 3D cell viability assay kit has a greater ability to lyse cells in spheroids than the 2D cell viability assay kit. Therefore, we hypothesized that the 3D cell viability assay kit would be more effective in our studies. In brief, in 2D assays, 50 μL of PBS was added to each well followed by the adding of 50 μL CellTiter-Glo reagent which was prepared according to the manufacturer’s protocol. To induce cell lysis, there contents were mixed for 2 min on a plate shaker. The samples were then incubated at room temperature for 10 min. Lysate were then transferred to a white opaque 96 well plate. Luminescent was measured using a luminometer (FlexStation III; Molecular Device, San Jose, CA, USA). For 3D assays, the plates were mixed strongly for 5 min to induce cell lysis and then incubated for 25 min at room temperature to allow the luminescence signal to stabilize. Finally, 50 μL of the solution was transferred to a white opaque 96-well plate to be measured with a luminometer as a relative luminescence unit with an integration time of 1 sec.
To measure enzyme leaking due to cellular damage, lactate dehydrogenase (LDH) activities were estimated using a LDH cytotoxicity detection kit (Takara Bio, Otsu, Japan) according to the manufacturer’s instruction. In brief, HepG2 and HepaRG cells were seeded on 96-well cell culture plates (1.5 × 104 cells/mL) with culture medium and exposed to hepatotoxicants. Following the 24 hr treatment period, it was accordance with the manufacturer’s instructions for use. The absorbance was read on a luminometer (FlexStation III) at 490 nm. Cell-free wells were used to eliminate background absorption from nanoparticles. The serum activity of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured using commercial kits specific for each assay. The manufacturer’s instructions were followed for the protocol. ALT levels were measured using ALT activity assay kit (Sigma-Aldrich). To the cell free wells containing the reaction mixture, serum samples were added. AST levels were measured using AST activity assay kit (Sigma-Aldrich). After incubated with reaction mix for 5 min at 37°C. Absorbance was read at a wavelength of 570 nm for ALT and 450 nm for AST on a luminometer (FlexStation III). Samples were measured for absorbance at 570 nm and 450 nm every 5 min for up to 30 min. After completing the measurement, the values were calculated by subtracting the starting measurement from the last measurement.
HepG2 and HepaRG cells were seeded into 96-well cell culture plates (1.5 × 104 cells/mL) with culture medium. On the following day, cells were treated with serial concentrations of ADR and AAP for 24 hr. To remove floating cells, the supernatants collected from each well were centrifuged at 3,000 × g for 5 min. Supernatants were then stored at –20°C until assay. For the estimation of albumin (ALB) and urea, respectively, an ALB Human ELISA kit (Abnova, Walnut, CA, USA) and a urea assay kit (Cell Biolabs, San Diego, CA, USA) were performed following the instructions of the respective supplier. Absorbance was read at a wavelength of 450 nm for ALB and 630 nm for urea on a luminometer (FlexStation III). ALB and urea levels were calculated using each standard curve. They were normalized according to cell count (1.5 × 104 cell/mL).
The RNeasy Mini Kit (Qiagen, Hilden, Germany) was used to extract RNA from the cells. The concentration and purity of RNAs were assessed with an ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). cDNA synthesis was performed using 1 μg of the total RNA with an Omniscript RT kit (Qiageny). Polymerase chain reaction (PCR) cycling were denaturation at 95°C for 10 sec, annealing at 45°C to 64°C for 10 sec, and extension at 72°C for 20 sec for up to 40 cycles. After reverse transcription of RNA into cDNA, expression levels of CYP1A2, CYP2C9, and CYP3A4 were measured by qPCR. A CFX Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) was used to amplify target gene using primers. The mRNA level of each test gene (CYP1A2, CYP2C9, and CYP3A4) was normalized to β-actin according to the following formula: △CT = CT (test gene) – CT (β-actin). Thereafter, relative mRNA level of each gene was calculated using the △△CT method: △△CT (test gene) = △CT (test gene in the treated sample) – △CT (test gene in the untreated sample). The fold change of mRNA level was expressed as the relative expression of 2–△△CT. Induction was defined as more than 2-fold over the vehicle control and 20% or more over the positive control. The sequences of primer sets are shown in Table 2.
GraphPad Prism software version 5.0 was used to analyze the experimental data by tow-way analysis of variance (ANOVA) followed by Bonferroni post-test. The data were considered to be significant at p<0.05 (*). GraphPad Prism software 5.0 was used to calculate the 50% inhibitory concentration (IC50) value (logarithmic transformation of X-values and nonlinear regression sigmoidal dose-response analysis with variable slope-with bottom and top constraints set at 0 and 100, resp.). Values are presented with ± 95% confidence intervals.
RESULTS
Cytotoxicities of ADR and AAP were evaluated based on cell viability using two different cell culture types (2D and 3D cultures) and compared with those of HepG2 and HepaRG cells without ADR or AAP treatment. ADR and AAP decreased cell viability of both cell lines and culture types in a dose-dependent manner (Fig. 1A and B). However, different responses were seen depending on the cell culture type. For both cells, cell viability was rapidly decreased in 2D culture, showing significant differences from 3D culture. The IC50 value of ADR was found to be 44.85 μM for HepG2 2D, 65.47 μM for HepaRG 2D, 55.64 μM for HepG2 3D, and 109.38 μM for HepaRG 3D. The IC50 value of AAP was determined to be 14.95 mM for HepG2 2D, 7.11 mM for HepaRG 2D, 48.43 mM for HepG2 3D, and 26.19 mM for HepaRG 3D (Table 3). Overall, 2D cultures were found to be more sensitive to drugs than 3D cultures.
| IC50 value | HepG2 2D | HepaRG 2D | HepG2 3D | HepaRG 3D |
|---|---|---|---|---|
| Amiodarone HCl IC50 (μM) | 44.85 | 65.47 | 55.64 | 109.38 |
| Acetaminophen IC50 (mM) | 14.95 | 7.11 | 48.43 | 26.19 |
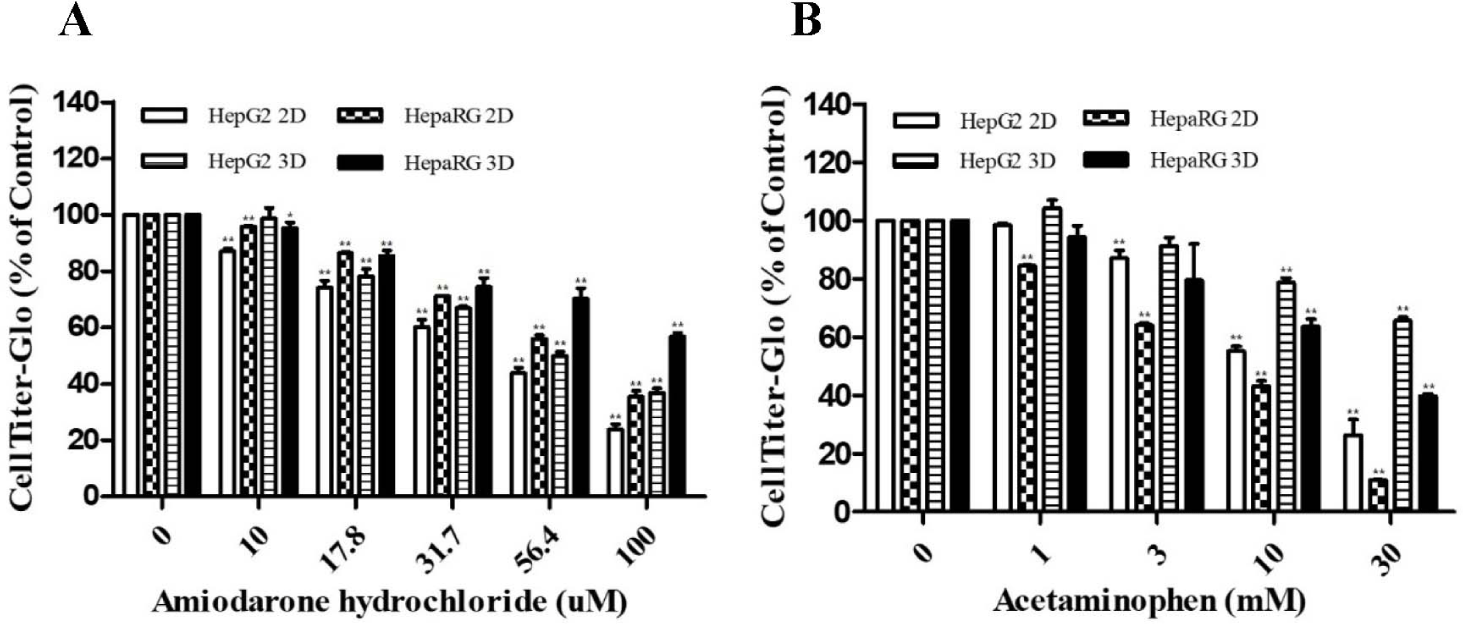
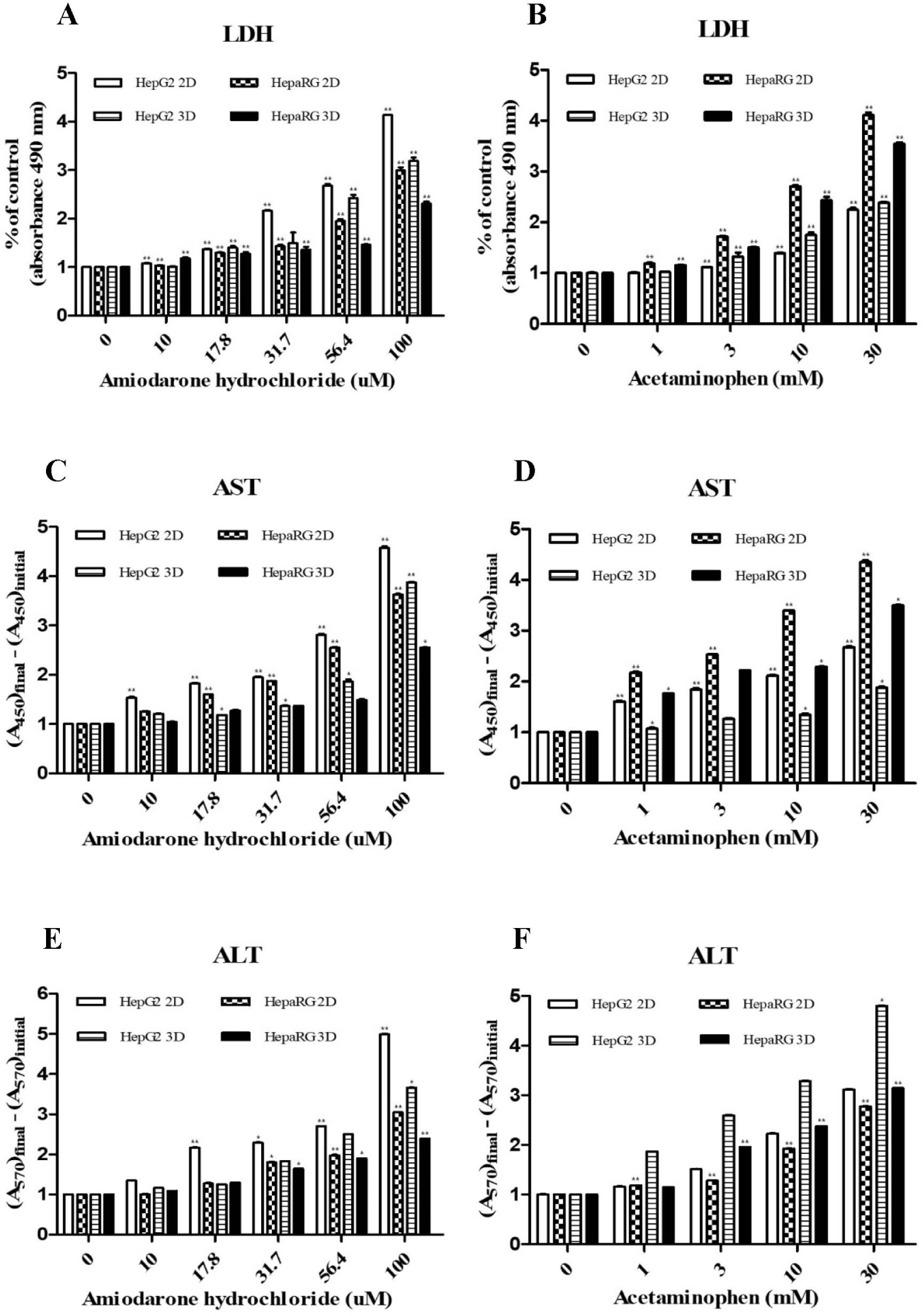
To measure the expression of LDH, AST and ALT (liver injury markers), HepG2 cells and HepaRG cells were cultured in 2D and 3D and then treated with hepatotoxicants ADR and AAP. Activities of LDH (Fig. 2A and B), AST (Fig. 2C and D), and ALT (Fig. 2E and F) were increased by treatment with ADR or AAP in a dose-dependent manner in both cell lines and culture types. However, different responses were seen depending on the type of cell culture. It was confirmed that their activities in both cells were increased more in 2D culture than in 3D culture in a drug concentration-dependent manner. HepG2 cells showed higher expression of liver injury factors after ADR treatment than HepaRG cells and 2D cultures showed higher expression of these factors than 3D cultures. HepaRG cells showed higher expression of these factors after AAP treatment than HepG2 cells and 2D cultures showed higher expression of factors than 3D cultures. These results were consistent with results of the cell viability assay.
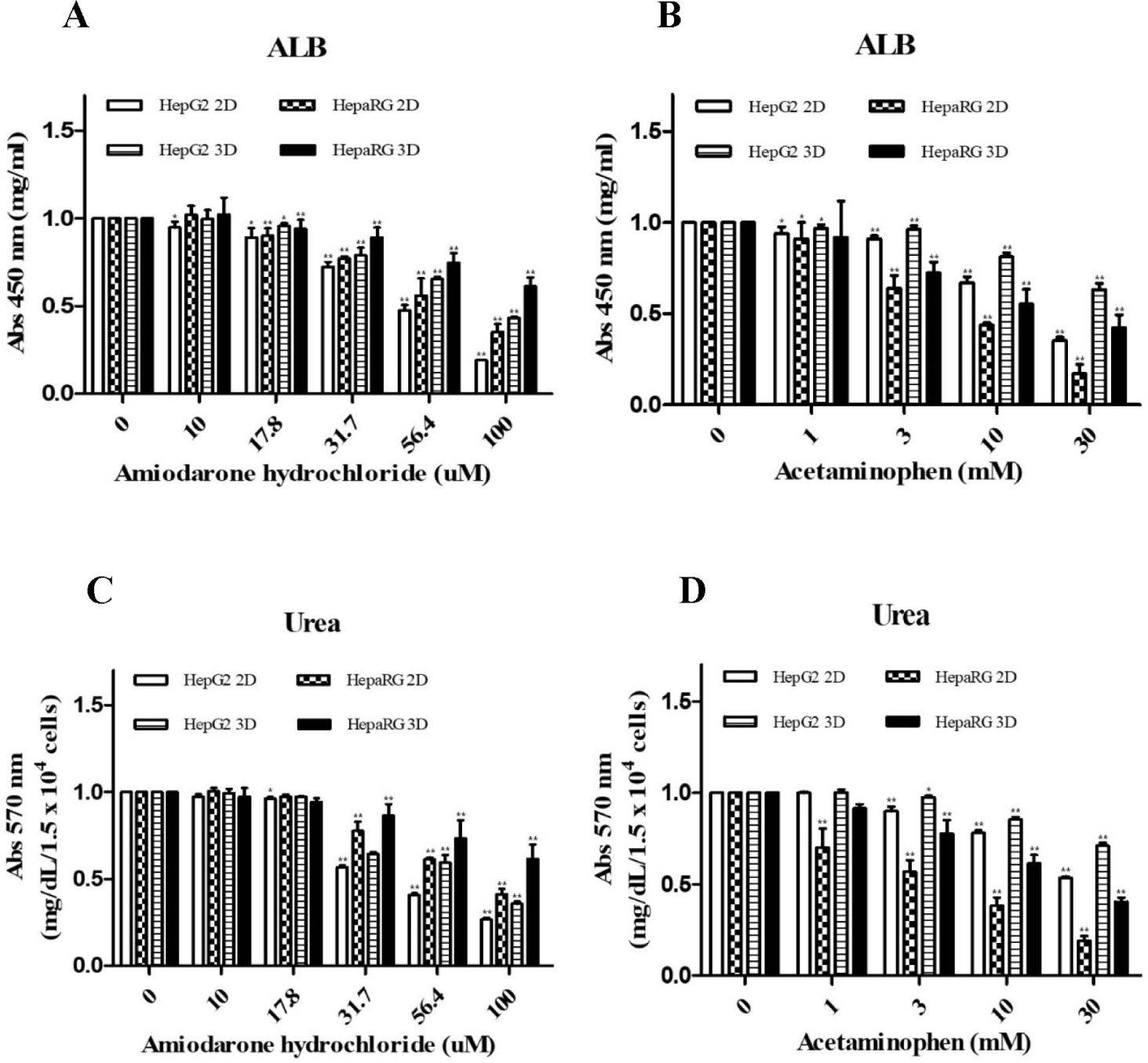
To determine effects of cell culture type and treatment with hepatotoxicants on expression levels of liver specific markers, differences in ALB and urea secretion between 2D and 3D cultures were analyzed. Each result was based on the control of HepG2 2D culture. Quantitative analysis of ALB showed that its expression levels were higher in 3D cultures and HepaRG cells than in 2D cultures and HepG2 cells, respectively. In addition, after culturing each cell, cells were treated with ADR or AAP to analyze changes in ALB and urea secretion depending on the drug concentration. As a result, it was found that ALB secretion was reduced depending on the concentration of each drug (Fig. 3A and B). Urea secretion analysis gave the same results as ALB secretion (Fig. 3C and D).
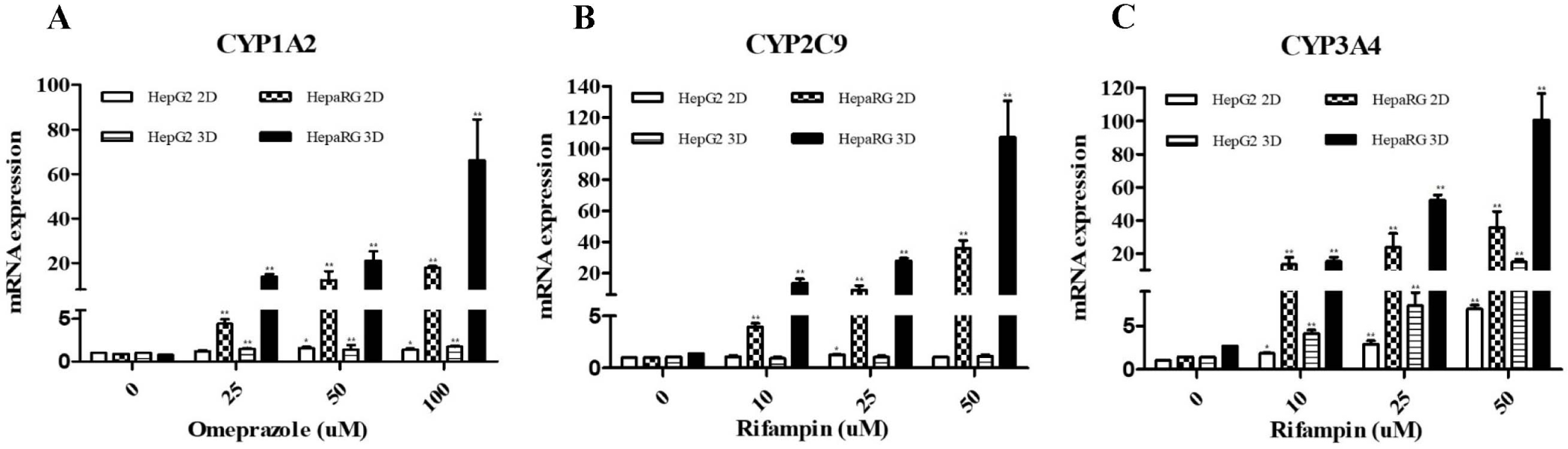
We treated each cell with a CYP enzyme inducer and then examined gene expression level of the enzyme to determine which cells and culture types had higher expression levels of the CYP450 enzyme known to be involved in drug metabolism in the liver. Each result was based on the control of HepG2 2D culture. Expression levels of CYP1A2, CYP2C9, and CYP3A4 enzymes in HepG2 and HepaRG cells were found to show significant differences after treatment with drugs. In HepaRG cells, expression levels of CYP1A2, CYP2C9, and CYP3A4 were increased after treatment with inducers. Compared with HepaRG cells, HepG2 cells had relatively lower levels of CYP1A2, CYP2C9, and CYP3A4. Additionally, significant differences were seen between 2D and 3D cultures. There was no difference in the expression of CYP1A2 and CYP2C9 in 2D and 3D cultures of HepG2 cells. However, CYP3A4 had higher expression levels in 3D cultures than in 2D cultures. In HepaRG cells, all three enzymes CYP1A2, CYP2C9, and CYP3A4 were found to have higher expression levels in 3D cultures than in 2D cultures. Activities of CYP1A2 in 2D and 3D cultures of HepaRG were significantly (p<0.05) increased after treatment with 25 μM of omeprazole (Fig. 4A). Activities of CYP2C9 and CYP3A4 were significantly (p<0.05) increased after treatment with 10 μM rifampin (Fig. 4B and C). In HepG2 cell 2D and 3D cultures, the activity of CYP3A4 was significantly (p<0.05) increased after treatment with rifampin at 10 μM (Fig. 4C). But, CYP1A2 and CYP2C9 were not increased (p<0.05) in 2D and 3D cultures of HepG2 cells (Fig. 4A and B).
DISCUSSION
Although the use of PHHs [21], their high-throughput in vitro drug screening is severely limited by their short lifespan, high cost, limited availability and observed inter donor variability. Therefore, the immortalized hepatic lines HepG2 and HepaRG were used in this study. While HepG2 cells have many limitations in 2D culture, including the loss of hepatic-specific structure and functionality [21], the ease of handling compared to PHHs, coupled with a stable phenotype and lack of donor variability [21., 22], offers many advantages for standard operating procedures development in preclinical drug screening. On the other hand, culturing HepG2 cells in a 3D environment has been shown to restore some of the characteristics lost when cultured in a 2D environment. In addition, 3D cultured HepG2 cells has a reduced proliferation rate, self-organize and differentiate into 3D spheroids that restore lost liver structure and function [23., 24]. HepaRG cells are a hepatocellular carcinoma-derived human cell line expressing functional liver markers such as several CYPs and phase II enzymes and nuclear receptors. In CYP3A4 induction studies, HepaRG cells have been shown to be an excellent surrogate for PHHs. Therefore, we cultured cells in 2D and 3D cell culture type using HepG2 and HepaRG cells, which are most commonly used for hepatotoxicity studies. We also compared the response to drug treatment by each cell and culture type. Cell viability was measured for both 2D and 3D spheroid viability using CellTiter-Glo® ATP quantification. In parallel with the assessment of viability by quantification of ATP, the LDH assays were used to determine the cytotoxicity induced by ADR and AAP. Because the LDH assay is supernatant based, the cytosolic enzyme LDH is released into the culture supernatant by compromised plasma membrane, avoiding the problems of dissociating the spheroid structure required by the other membrane consistency methods such as neutral red. When evaluated using these assays, we observed decreased viability or increased cytotoxicity induction after treatment with ADR and AAP in both culture types. When comparing the responsiveness to the drugs in each cell type, HepG2 cells were more sensitive to ADR treatment, while HepaRG cells were more sensitive to AAP treatment. Changes in cell viability based on cell culture type were also observed. For both HepG2 and HepaRG cells, it was observed that 2D cultures were more sensitive to drugs than 3D cultures. These results were also observed as a result of cytotoxicity induction. The expression of LDH increased in a drug concentration-dependent manner, with 2D cultures being more sensitive than 3D cultures. AST and ALT are markers of liver injury that increase when cells are damaged [25]. Two well-known markers of liver function are ALB and urea. ALB is a major protein synthesized in the liver and plays an important role in maintaining tumor pressure, transporting various molecules and regulating pH balance. Urea is a waste product formed in the liver during protein metabolism. The liver׳s ability to metabolize ammonia into urea is crucial for maintaining nitrogen balance in the body [26., 27]. HepG2 and HepaRG cells were cultured in 2D and 3D forms and each cell was treated with ADR and AAP to determine the extent of liver damage. Similar to cell viability and toxicity tests, the expression of AST and ALT increased in a drug concentration-dependent manner. Similar results were observed for the secretion of ALB and urea. Secretion decreased in a concentration-dependent manner and expression was significantly reduced in 2D cultures. In addition, HepaRG cells had higher basal expression compared to HepG2 cells when tested under the same conditions. In the same cells, expression of ALB and urea was higher in cells cultured in a 3D cultures. The hepatotoxicants ADR and AAP cause mitochondrial dysfunction [28] and liver failure [29]. ADR and AAP has been reported to cause hepatotoxicity and decrease cell viability in HepG2 [30., 31] and HepaRG [32., 33] cells. These findings are consistent with our results which showed decreased cell viability, increased expression of liver injury markers, and decreased liver function after drug treatment. The evaluation of DILI using hepatocytes relies on an accurate understanding of the characteristics of the cells and drugs, which is difficult to achieve. There are studies that show different liver cells have different responses to the same drug [34]. Alternatively, studies have shown that when a single cell is treated with different drugs and shows varying sensitivity to each drug [17]. Therefore, it is important to study liver toxicity using a variety of cells and drugs, as we have done in our study. A number of critical enzymes play an important role in cellular metabolism under disease conditions or in the metabolism of drugs. CYPs have been extensively studied since their identification because of their involvement in the metabolism of a wide range of substrates, including components of biological processes and therapeutic agents, in the human body [31., 35]. In particular, it is reported that CYPs catalyze over 95% of oxidative and reductive reaction [31]. They activate or inactivate exogenous substrates such as drugs, food additives, endogenous compounds like steroids, xenobiotics and fatty acids [36.-38]. In particular, CYP1A2, CYP2C9 and CYP3A4 enzymes are known to be the most involved in drug metabolism in human hepatocytes. CYP1A2 is a member of the CYP family primarily found in the liver, although it is also present in other tissues. CYP1A2 is responsible for the metabolism of various substrates, including caffeine, theophylline, certain drugs, and environmental toxins like polycyclic aromatic hydrocarbons found in cigarette smoke. CYP1A2 has been extensively studied due to its role in drug metabolism and its potential impact on drug efficacy and toxicity [39]. CYP2C9 is another important CYP predominantly expressed in the liver, although it is also found in other tissues. CYP2C9 substrate specificity and catalytic properties have been extensively studied to understand its role in drug metabolism and pharmacokinetics [40]. CYP3A4 is one of the most abundant CYPs in the liver and intestine, playing a pivotal role in the metabolism of a wide range of drugs. It metabolizes approximately 50% of clinically used drugs, including statins, immunosuppressants, antivirals, and many others. Due to its broad substrate specificity and significant contribution to drug metabolism, CYP3A4 is a major determinant of drug clearance and efficacy [41]. In this study, we compared the mRNA expression of CYP enzymes in HepG2 and HepaRG cells in 2D and 3D cultures. The expression of each enzyme was induced by treating the cells with known standard inducers. HepG2 cells showed no expression of CYP1A2 and CYP2C9 enzymes in both 2D and 3D cultures. On the other hand, the expression of CYP3A4 was increased by inducer treatment. It also showed higher expression in 3D culture. As demonstrated in various studies, HepG2 cells normally express CYP1A2 [42]. However, there is evidence that while caffeine induces CYP1A2 expression in rat hepatocytes, it may not have the same effect in human HepG2 cells [43]. HepG2 cells generally express CYP1A2, but the expression level and regulatory factors can vary depending on the experimental conditions and stimuli. There is also evidence that HepG2 cells have low expression of CYP2C9, an important enzyme involved in drug metabolism [44.-46]. This low expression affects their suitability as a model for studying hepatotoxicity and drug metabolism [47., 48]. It is important to consider this limitation when using HepG2 cells as a model system for drug metabolism research. In contrast to HepG2 cells, HepaRG cells showed expression of CYP1A2, CYP2C9 and CYP3A4 enzymes in both 2D and 3D cultures. Furthermore, we found that all three enzymes were more highly expressed in 3D cultures than in 2D cultures. Interestingly, the expression of all enzymes was much higher than in HepG2 cells. HepaRG cells are a widely used model for drug metabolism studies due to their ability to express a variety of CYPs [49]. They respond to typical inducers by inducing a variety of CYPs, including CYP1A2, CYP2C9, and CYP3A4 enzymes, which are essentially involved in human drug metabolism [50]. HepaRG cells are known to be metabolically similar to human adult hepatocytes, making them useful for drug metabolism and toxicity studies [51]. These findings corroborate our results that HepaRG cells have a higher capacity to induce CYP enzymes compared to HepG2 cells. Furthermore, our results are consistent with studies showing increased mRNA levels of CYP1A2, CYP3A4 [52., 53] and increased expression of drug metabolizing enzymes including CYP2C9 in HepaRG 3D cultures [54]. In conclusion, the development of in vitro models that can predict DILI at the preclinical stage for drug development is becoming increasingly important. Therefore, the selection of in vitro models that can study a clearer and more diverse range of drug responses is important. In this study, HepG2 cells and HepaRG cells, which are often used in drug screening were compared, and each cell was cultured in 2D and 3D forms to evaluate them as in vitro models for drug development. The results of the study showed that the sensitivity of the cells varied depending on the type of cell, culture type and type of drug. In addition, HepaRG cells with higher expression of CYP enzymes involved in human drug metabolism are more suitable for drug metabolism evaluation, and 3D culture is more suitable as a model for drug metabolism evaluation than 2D culture. However, the results of CYP enzymes need to be validated by further experiments on metabolic mechanisms through enzyme activity measurement or liquid chromatography-tandem mass spectrometry metabolite.