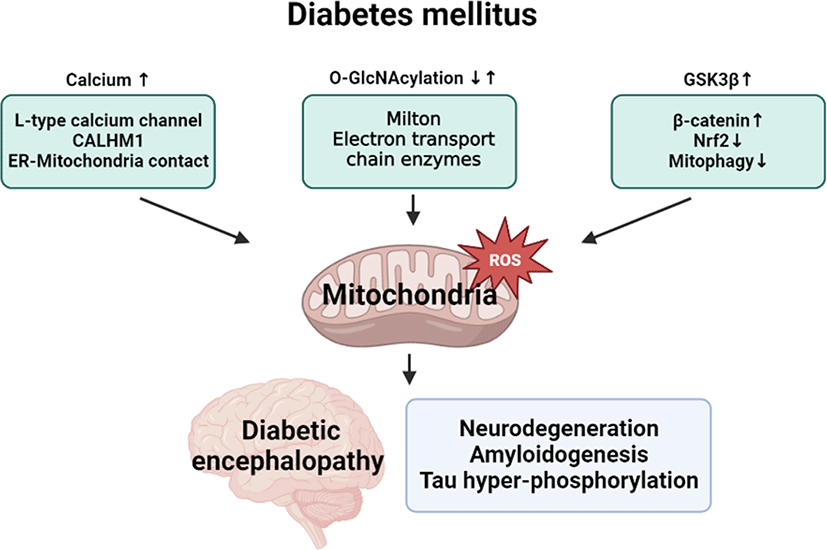
General introduction of diabetic encephalopathy
Diabetic encephalopathy is a chronic complication with neurodegenerative disease in patients with type 1 or type 2 diabetes mellitus (DM) who have been receiving long-term treatment. In 2005, Eric steen et al. described that the dysregulation of insulin and insulin-like growth factor (IGF) causes dementia such as Alzheimer’s disease (AD) and first proposed diabetic encephalopathy as type 3 diabetes [1]. A previous follow-up study investigating the relationships between blood glucose levels and dementia risk confirmed that a higher blood glucose level is a potential risk factor for dementia. Furthermore, the risk of developing dementia was found to be closely associated with high blood glucose levels without DM [2]. An epidemiological study showed that the number of patients with type 2 DM or dementia is strongly increasing and suggested DM as a risk factor for vascular dementia and AD [3]. Several studies using human cell models and animal models demonstrated that both type 1 DM and type 2 DM accompanied neurodegenerative phenotypes and dementia with cognitive impairment [4]. There is no clear information on a precise physiological mechanism underlying the development of diabetic encephalopathy in patients with DM. Nevertheless, the chronic inflammation, oxidative stress, vascular damage, protein dysfunction, low acetylcholinesterase activity, low growth factor levels, and metabolic abnormality occurring during the development of type 2 DM are involved in the occurrence of diabetic encephalopathy [5, 6]. For diabetic encephalopathy treatment, correcting the low acetylcholinesterase activity and oxidative stress was found to be an effective strategy for treating memory loss impairment in a streptozotocin (STZ)-induced mice model. All abbreviations used in this article are listed in Table 1.
Glucose transporter (GLUT) in diabetic encephalopathy
Previous studies have demonstrated that alterations in GLUT and glucose metabolism in patients with DM are the primary factors for AD pathology [7, 8]. Among GLUTs, both GLUT1 and GLUT3 contribute to glucose uptake in the brain. GLUT1 is primarily expressed in glial and endothelial cells, whereas GLUT3 is primarily expressed in neuronal cells [9]. Neuronal GLUT3 expression was found to be decreased in an intracerebral glucose concentration–dependent manner, but astrocytic GLUT1 expression was independent of intracerebral glucose levels [8]. A noninvasive in vivo imaging analysis demonstrated decreased glucose uptake in the brain of rat exposed to an intracerebroventricular injection of amyloid beta [10]. In a Drosophila AD model, GLUT1 overexpression was found to restore glucose uptake, which prolonged life span with neuroprotection through the suppression of proteostasis [7]. Another study showed that GLUT1 deficiency in the blood–brain barrier accelerated neurodegeneration through microvascular degeneration, amyloid beta deposition, and cognitive impairment [11]. In astrocytes, a hyperglycemic condition was found to impair membrane localization of GLUT1, decreasing the glucose bioavailability of astrocytes in an AD model [12]. Considering that glucose uptake and protein glycosylation were increased in high glucose–exposed cells and DM models, further investigation of GLUT regulation response to hyperglycemic conditions is required to clarify the role of GLUTs in diabetic encephalopathy.
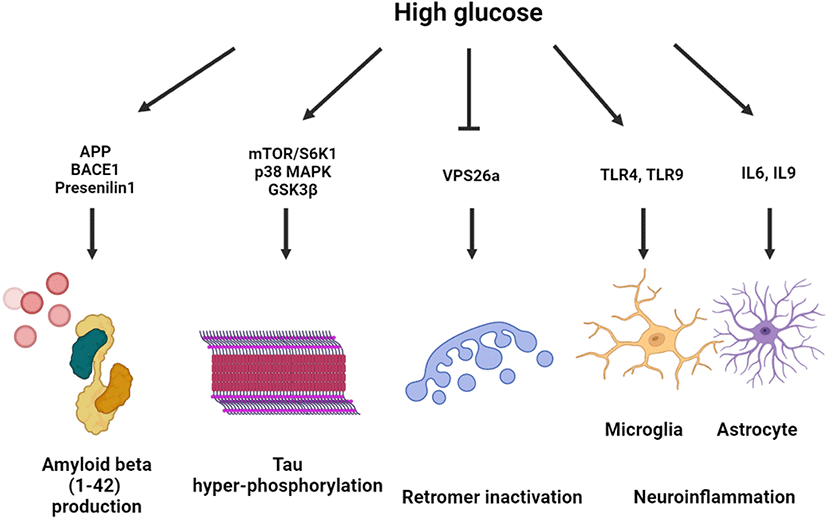
Effect of high glucose on neurodegeneration
High glucose–exposed neuronal and nonneuronal cells exhibited altered amyloid beta processing mechanisms through upregulated amyloid beta precursor protein (APP) gene transcription, beta-secretase 1 (BACE1)-mediated APP processing, and downregulated APP degradation [13, 14]. High glucose–activated Akt/glycogen synthase kinase 3β (GSK3β) pathway increased the expression levels of amyloid beta 1–42 and APP-cleaving enzymes, including BACE1 and presenilin 1, which was reversed by melatonin treatment [15]. APP processing under hyperglycemic conditions is controlled in a lipid raft–dependent manner. In neuronal cell line and primary hippocampal neuronal cells, it was observed that high glucose facilitated lipid raft reorganization and APP endocytosis of lipid raft with early endosomal enlargement [14, 16]. Furthermore, high glucose–induced DNA methyltransferase 1-dependent hypermethylation of vacuolar protein sorting-associated protein 26a (VPS26a), which interacts with the BACE1 enzyme. The induction of VPS26 was found to inhibit APP retention in the endosome, which subsequently suppressed amyloid beta and phosphorylated tau levels in neuronal cells [17].
Tau-regulated axonal transport contributes to GLUT4 trafficking, determining insulin resistance in the brain [18]. Moreover, high glucose conditions are closely associated with tau hyperphosphorylation and aggregation, forming neurofibrillary tangles (NFTs). It has been shown that high glucose decreased the expression of the lipid raft caveolae-forming protein Cav-1. Overexpression of Cav-1 inactivated the mammalian target of rapamycin (mTOR)/ribosomal protein S6 kinase beta-1 pathway, suppressing the high glucose–induced tau hyperphosphorylation [19]. Another study showed that toll-like receptor (TLR) 9-activated neuroinflammatory response under diabetic conditions increases tau hyperphosphorylation and neuronal apoptosis through p38 mitogen-activated protein kinase (MAPK) activation [20]. The dimethylbiguanide metformin, an antidiabetic drug, activated protein phosphatase 2a dephosphorylated hyperphosphorylated tau in direct and indirect mTOR-dependent manners [21]. Furthermore, protein glycosylation and S-nitrosylation (SNO) are involved in insoluble NFT formation and neurodegeneration. In a postmortem study, aberrant O-linked-N-acetylglucosaminylation (O-GlcNAcylation) signaling activation was detected in the brain of patients with AD [22]. O-glycosylation of tau at Ser400 under high glucose conditions was found to suppress the hyperphosphorylation of tau at Ser396 and Ser404 [23].
It has been reported that a hyperglycemic condition increases neuroinflammation in type 1 and type 2 DM. An astrocytic cell line exposed to high glucose exhibited increased inflammatory responses with an upregulated secretion of interleukin (IL)-6 and IL-8 [24]. Microglial activation with inflammatory gene upregulation was detected in animal models of both type 1 and type 2 DM [25]. In a BV2 microglial cell line, it was found that high glucose augmented lipopolysaccharide (LPS)-induced TLR4 expression and neuroinflammation [26]. In addition, p38 MAPK inhibition by sesamin treatment prevented from BV2 microglial cell activation and microglia-mediated neurotoxicity [27]. It was also observed that high glucose–stimulated triggering receptor expressed on myeloid cells 2 increased proinflammatory cytokine secretion and NLRP3 inflammasome activation, which play a key role in microglial activation in DM [28]. Collectively, high glucose exacerbates neurodegeneration through amyloidogenesis, hyperphosphorylation, retromer inactivation and neuroinflammation (Fig. 1).
Mitochondrial oxidative stress in diabetic encephalopathy
The brain is a high-oxygen-demand organ and sensitive to lipid peroxidation induced by oxidative stress [29]. An imbalanced intracellular redox status and dysfunction of the reactive oxygen species (ROS) antioxidant system play a major role in the oxidative stress of neuronal cells. The endogenous source of ROS is classified into nonmitochondrial and mitochondrial. In particular, mitochondrial ROS is considered as a major endogenous source increasing the ROS of neuronal cells [29]. It has been proposed that there are multiple pathways of mitochondrial ROS generation, which are controlled by mitochondrial respiratory chain complexes such as complex 1 and complex 3 [29]. A recent study described about mitochondrial disease, indicating mitochondrial pathogenesis due to diabetic encephalopathy [30]. Mitochondrial diabetes in diabetic encephalopathy includes mitochondrial DNA mutation, mitochondrial dysfunction, alterations in the expression of axonal transport proteins, and decreased mitochondrial membrane potential [30]. Therefore, several studies have focused on mitochondrial ROS–based preventive and therapeutic strategies for neurodegeneration, which emphasizes the therapeutic potential of mitochondrial regulatory drug development for diabetic encephalopathy.
Cellular ROS is catalyzed and removed by intracellular antioxidant enzymes and small-molecule antioxidants. Mitochondrial superoxide (SOD) inactivates oxygen radicals, forming hydrogen peroxide, which is further catalyzed by glutathione peroxide, catalase, and peroxiredoxin [31]. It has been well documented that oxidative imbalance plays a key role in AD pathogenesis with neuronal cell damage [32]. During the early phase of AD pathogenesis, the intracellular accumulation of amyloid beta elicits mitochondrial dysfunction with failure of energy production metabolism [33]. Moreover, increased intracellular ROS generation induced by mitochondrial dysfunction in AD was found to mediate amyloid beta production and tau hyper-phosphorylation, forming NFTs [33]. Cellular amyloid was commonly detected in the extracellular region, but it was also detected in intracellular organelles, including the Golgi apparatus, endoplasmic reticulum, and mitochondria. In particular, amyloid accumulation in mitochondria affects mitochondrial respiratory function and mitochondrial ROS levels [34]. In addition to neurodegeneration, a recent study demonstrated that mitochondrial stress also activated neuroinflammation in neurons and astrocytes [35]. These findings implicate that mitochondrial oxidative stress is a critical factor for the vicious cycle in neurodegeneration. Based on the therapeutic potential of mitochondrial oxidative stress, several studies have reported natural mitochondrial antioxidant drugs, such as berberine and avocado oil, as potential drug candidates for treating diabetic encephalopathy through the restoration of mitochondrial function [36, 37].
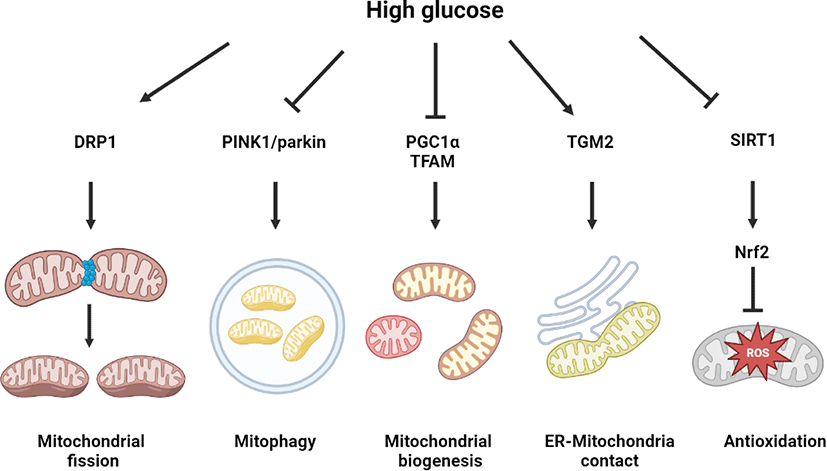
Effect of high glucose on mitochondrial dynamics and nuclear factor erythroid 2–related factor 2 (Nrf2)-regulated antioxidative system
Mitochondrial dynamics change is a representative phenotype of cells exposed to high glucose conditions, associated with various diabetic complications, including diabetic encephalopathy, diabetic retinopathy, diabetic nephropathy, and diabetic heart. In a metabolomics study on an STZ-induced type 1 DM rat model, aberrant alterations of mitochondrial dynamics induced cognitive impairment in diabetic encephalopathy [38]. N-methyl-D-aspartate receptor (NMDAR) activation by high glucose was found to increase calcium and nitric oxide (NO) levels for the SNO of insulin-degrading enzyme and dynamin-related protein (DRP), which mediated mitochondrial dysfunction and neurodegeneration, including synaptic damage, dendritic loss, and cognitive impairment [39]. Other studies showed that in retinal cells, endothelial cells, and cardiomyocytes, high glucose–activated mitochondrial fission, inducing a fusion–fission imbalance and mitochondrial dysfunction through AMP-activated protein kinase (AMPK) inactivation and DRP1 activity regulation [40–42]. In podocytes, high glucose inhibited PGC1α- and TFAM-dependent mitochondrial biogenesis and PTEN-induced kinase 1 (PINK1)-dependent mitophagy, as well as mitochondrial fission [43]. In neuronal cells, high glucose primarily upregulated the PINK1–parkin pathway for preventing mitophagy from excessive oxidative stress generation [44]. Melatonin-induced melatonin receptor type 2 (MT2) signaling was found to enhance PINK1–parkin pathway-dependent mitophagy, which is effective in preventing high glucose–induced neuronal cell death [44]. Mitophagy activation by parkin overexpression was found to activate DRP1-mediated mitochondrial fission, which alleviated the high glucose–induced apoptosis of cardiomyocytes [45]. This finding indicates a mutual positive interaction between mitophagy and mitochondrial fission. Under high glucose conditions, neuronal cells have high levels of mitochondrial calcium with endoplasmic reticulum (ER)–mitochondrial contact. High glucose–induced the expression of transglutaminase 2, mediating the interaction between voltage-dependent anion channel 1 and IP receptor for forming the ER–mitochondrial contact [46]. Urolithin A, a microbiome-derived metabolite, was reported as an ER–mitochondrial contact inhibitor, which reduced mitochondrial calcium levels, alleviating mitochondrial oxidative stress in neuronal cells under high glucose conditions [46].
The Nrf2-regulated antioxidant system has a potent capacity of mitochondrial ROS suppression in neuronal cells [47]. In the mouse hippocampal neuron cell line HT22, high glucose–induced ROS generation with Nrf2 and heme oxygenase 1 suppression. Treatment with Nrf2 activator suppressed the high glucose–induced neuronal cell death [47]. In rat retinal cells, it was found that high glucose decreased the expression of Nrf2 target genes, including glutathione and catalase with redox regulatory capacity [48]. In Nrf2 knock down and knock out assays, it was demonstrated that Nrf2 is a major protective factor for high glucose–induced mitochondrial oxidative stress and cellular apoptosis [49]. It has been well documented that sirtuin 1 (SIRT1) is a critical regulator of Nrf2 activity. It was found that activation of the salvianolic acid–induced SIRT1–Nrf2 pathway suppressed oxidative stress and neuronal degeneration [50]. High glucose–induced SIRT1 activation reduced neuronal apoptosis through p53 deacetylation in STZ-induced diabetic mice and SH-SY5Y neuroblastoma cell line [51]. Moreover, SIRT1 has been reported as a suppressive factor of insulin resistance, which is closely associated with SIRT1-regulated mitochondrial respiratory chain activity. In fact, SIRT1 overexpression was found to restore the activity of high glucose–reduced mitochondrial complex 1, which was reversed by SIRT3 knock down [52]. Nevertheless, there is a need for further investigation of the detailed mechanism by which high glucose regulates SIRT1 activity related to mitochondrial function and Nrf2 pathway to develop SIRT1-dependent therapeutic strategies for diabetic encephalopathy. Collectively, high glucose regulates mitochondrial dynamics and 2).
Role of calcium in diabetic encephalopathy
Calcium homeostasis in brain tissue has been considered to be the major cause of diabetic encephalopathy [53]. Several studies have demonstrated that intracellular Ca2+ levels were increased in the brain of patients with diabetic encephalopathy, and large amounts of calcium inflow cause calcium homeostasis disorder in the brain [54]. Intracellular calcium is maintained by store-operated calcium entry (SOCE), the process by which emptying of ER calcium stores causes the influx of calcium across the plasma membrane for calcium signaling. Disorders related to SOCE cause the dysregulation of Ca2+ signaling in neuronal cells [55]. The L-type calcium channel (LTCC) is a plasma membrane–localized voltage-dependent calcium channel and is distributed in neuronal cell bodies [56]. LTCC has four isoforms, and CaV1.2 expression was increased in diabetic neurodegeneration by excessive calcium absorption [57]. Hence, the dysregulation of LTCC has been reported in various neurodegenerative diseases, including AD [57]. Nimodipine, an LTCC blocker, improves diabetic encephalopathy by blocking calcium channels, reducing the expression level of CaV1.2 and absorption of mitochondrial Ca2+ [57]. Calcium/calmodulin-dependent protein kinase IV (CaMKIV) activated by calmodulin is responsible for regulating the synthesis and release of neurotransmitters, and an increase in Ca2+ levels induces the activation of CaMKIV, inhibiting synaptic formation and resulting in neuronal loss and several neurodegenerative disorders, including AD and Parkinson’s disease [58, 59]. The calcium homeostasis modulator 1 channel (CALHM1) is a voltage-gated channel that increases cytosolic calcium influx in response to decreases in extracellular calcium levels. The role of CALHM1 in neurodegenerative diseases remains unclear, and a polymorphism has been detected in the genome of patients with AD, which was expected to disrupt calcium homeostasis [60]. Therefore, the impairment of calcium signaling or calcium channel regulation causes neurodegenerative pathogenesis under DM conditions, resulting in diabetic encephalopathy.
Calcium signals are involved in various functions of mitochondria and mtROS regulation. Mitochondrial Ca2+ absorption affects the tricarboxylic acid (TCA) cycle and electron transfer chain (ETC) activity to regulate ATP production and generate ROS signals [61]. Excessive calcium levels stimulate respiratory chain activity, leading to higher amounts of ROS [62]. Mitochondrial Ca2+ directly stimulates mtROS-producing enzymes such as glycerol phosphate and α-ketoglutaric acid dehydrogenase to form mtROS [62]. Ca2+ induces the generation of NO through a regulator of NO synthases, and the generated NO can block complex IV, causing mtROS generation [63]. Furthermore, the continuous calcium load opens the pathway in the inner mitochondrial membrane, ruptures the outer mitochondrial membrane, and reduces the mitochondrial membrane potential [64]. The reduced mitochondrial membrane potential also causes the generation of ROS and the generation of more Ca2+ levels, separating cytochrome c from the inner membrane cardiolipin. Eventually, mitochondrial permeable transition pores are opened, and the apoptosis factor Ca2+ is released into the cytoplasm. Calcium homeostasis in mitochondria is closely related to neurodegenerative diseases. Recent studies have reported mitochondrial calcium uniporter (MCU) as a candidate treatment target for neurodegenerative diseases through mitochondrial Ca2+ regulation [65]. It has been reported that TG-2112x, the MCU inhibitor, can partially inhibit mitochondrial calcium absorption and protect neurons [66]. However, ROS produced by excessive calcium increases the release of more calcium to the mitochondria through the calcium channel between ER and the mitochondria, and calcium is transferred to the mitochondrial substrate through the MCU. MCU oxidation by ROS induces the S-glutathionylation of the Cys-97 residue of the MCU protein and exhibits continuous MCU channel activity with higher Ca2+ absorption in the mitochondria, increased mitochondrial ROS generation, and improved mitochondrial Ca2+ overload–induced apoptosis [67]. Moreover, high glucose–induced ER–mitochondrial contact was found to increase mitochondrial Ca2+ accumulation, which is critical for the pathogenesis of diabetic encephalopathy. Urolithin A was found to disrupt the ER–mitochondrial contact and alleviate neurodegeneration [46]. Excess Ca2+ continuously activates the respiratory chain and calcium channels of mitochondria, resulting in mitochondrial ROS generation and neurodegeneration. Therefore, inhibition of mtROS production through the regulation of calcium homeostasis is a potential treatment for diabetic encephalopathy.
Role of O-linked-N-acetylglucosaminylation (O-GlcNAcylation) in diabetic encephalopathy
O-GlcNAcylation is a posttranslational modification process that plays a vital role in signaling and biogenesis metabolism [68]. Increased O-GlcNAcylation causes mitochondrial ROS induction and decreases ATP production and membrane potential, resulting in dysfunction and inflammatory reactions of mitochondria and apoptosis under hyperglycemia. Increased O-GlcNAcylation of TCA cycle enzymes and ETC complexes was found to increase ATP production but reduce ROS production in mouse mitochondria [69]. However, another study confirmed that the rate of O-GlcNAcylation in the human body was increased under hyperglycemia [70]. Excessive O-GlcNAcylation of mitochondrial proteins inactivated ETC complexes, leading to mitochondrial dysfunction in cardiomyocytes under hyperglycemia [71]. Increased O-GlcNAcylation of calcium/CaMKII in ventricular myocytes was found to increase calcium leakage and limit reabsorption, resulting in arrhythmia in diabetic hyperglycemia [72]. Moreover, increased O-GlcNAcylation of COXI and NDUFA9 protein was found to reduce ATP production and mitochondrial membrane potential, causing mitochondrial damage by generating ROS [73]. Increased O-GlcNAcylation in vascular endothelial cells under hyperglycemia was due to reduced expression of the microRNA 200a/200b family, which inhibited OGT expression. Increase of O-GlcNAcylation by OGT was found to elevate the expressions of intercellular adhesion molecular 1, vesicular cell adhesion molecular 1, and e-selectin, resulting in endothelial inflammation [74]. Conversely, an increase of O-GlcNAcylation in neuronal cells under high glucose conditions was also observed. It was found that increased O-GlcNAcylation of milton, the mitochondrial motor protein, caused a reduction of mitochondrial transport to the synapse in mouse neurons under hyperglycemia [75]. Moreover, upregulated O-GlcNAcylation of the AMPA receptor subunits GluA1 and GluA2 mediated the NMDAR-dependent long-term potentiation and long-term depression in the hippocampal synapse [76]. Nonetheless, the role of O-GlcNAcylation in diabetic encephalopathy and its relationship with mitochondrial ROS has not yet been clarified.
It has been reported that chronic hyperglycemia causes insulin resistance in the brain, resulting in reduced O-GlcNAcylation in patients with AD [77]. Reduced O-GlcNAcylation of milton was found to cause the mislocalization of mitochondria in neuronal cells. Subsequently, ATP and Ca2+ supply was impaired, which hindered synaptic formation [78]. Because the tau protein has more serine/threonine phosphorylation sites, where O-GlcNAcylation occurs, reduced O-GlcNAcylation in the AD brain with impaired glucose metabolism is closely associated with tau pathology, including the hyperphosphorylation of tau protein [79]. Therefore, the regulation of O-GlcNAcylation is an extremely important approach for AD treatment. O-GlcNAcase is a potential treatment for recovering the reduced O-GlcNAcylation in the brain of patients with AD. Treatment with the O-GlcNAcase inhibitor thiamet-G reduced the separation of proteins from O-GlcNAc by inhibiting the function of OGA. Treatment with thiamet-G also improved the membrane potential or ATP production function of mitochondria in the brain [78]. Moreover, increased O-GlcNAcylation of tau protein was found to inhibit hyperphosphorylation, suppressing the formation of NFTs [80].
Role of glycogen synthase kinase 3β (GSK3β) signaling in diabetic encephalopathy
In DM, GSK3β is a vital signaling molecule in glycogen synthesis and blood glucose control, and recent studies have demonstrated that the GSK3β signaling pathway is closely related to diabetes and neurodegeneration. When insulin and insulin receptors bind each other, phosphoinositide 3-kinases (PI3K)/Akt signaling inhibits the phosphorylation of GSK3β for tau dephosphorylation. In diabetes, insulin resistance was found to impair the activation of the PI3K/Akt pathway, causing GSK3β activation and oxidative stress generation [81]. Similarly, the dysfunction of insulin or IGF-1 signaling system eventually stimulated GSK3β by blocking the PI3K/Akt/Wnt signaling pathway [82]. It was found that Wnt3a-inhibited GSK3β induced β-catenin accumulation for TCF activation, which enhanced the production of NeuroD1-stimulated insulin [83].
GSK3β activation directly promotes neurodegeneration through tau hyperphosphorylation, neuroinflammation, and neuronal dysfunction. It has been reported that GSK3β is involved in synaptic plasticity and hippocampal long-term potentiation, a mechanism essential for memory formation [84]. Another study showed that patients with AD exhibited upregulated GSK3β activity, suggesting that hyperactivation of GSK3β is an early event that precedes and accompanies the formation of NFTs and other tau-positive inclusions [85]. There is also extensive research showing that hyperactivated GSK3β signaling is critical for amyloidogenesis and microglial activation in AD pathogenesis [84]. In fact, several GSK3β inhibitors, including lithium chloride, are currently being investigated in preclinical and clinical trials for AD due to their anti-neurodegeneration properties [86].
The GSK3β signaling pathway is essential for mitochondrial oxidative stress and mitochondrial regulation. GSK3β-inhibited β-catenin causes mitochondrial damage due to oxidative stress, which is critical for the pathogenesis of diabetic retinal degeneration. This finding indicates that active β-catenin signaling can be a diagnostic biomarker for the early onset of diabetic retinopathy [87]. IGF-1-mediated GSK3β inactivation promoted mitochondrial biogenesis through upregulation of the transcriptional coactivators peroxisome PGC1β and PGC1-related coactivator. Furthermore, the IGF-1/GSK3β pathway plays a key role in Bcl-2 interacting protein 3-dependent mitophagy and Nrf2 stabilization, involving in mitochondrial ROS homeostasis [88]. In addition, the Akt/GSK3β-regulated signal transducer and activator of transcription 3 (STAT3) plays an essential role in mitochondrial function and oxidative stress caused by DM. STAT3-Cyp-D interaction plays an essential role in regulating ROS generation and mitochondrial function by regulating mitochondrial permeability transition pores, ATP production, and ETC. This finding indicates that inhibition of the GSK3β-STAT3 pathway is a potential strategy for improving mitochondrial function and anti-apoptosis [82]. In the SH-SY5Y cell line, AMPK activation stimulated GSK3β signaling, inducing the activation of mitochondrial transcription factor A, which leads to the transcription/translation of mitochondrial DNA, promoting mitochondrial biosynthesis [89]. Considering that GSK3β acts as a key player linking between DM and neurodegeneration, GSK3β-based therapeutic strategies are promising for the prevention and treatment of diabetic encephalopathy.
Conclusion
Although the respective role of mitochondrial oxidative stress in DM and neurodegeneration have been well documented, there is a lack of a clear understanding of the detailed relationships between DM condition and diabetic encephalopathy pathogenesis. For developing preventive and therapeutic strategies of diabetic encephalopathy in DM patients, it is necessary to further investigate mitochondrial regulatory mechanism in neuronal cells under hyperglycemic condition. Finally, we suggest mitochondrial oxidative stress regulatory targets in diabetic encephalopathy, such as calcium, O-GlcNAcylation, and GSK3β signaling (Fig. 3).