INTRODUCTION
Aralia elata (Araliaceae), which possesses various pharmacological activities including anti-arrythmia, antitumor, anti-inflammatory, anti-oxidant and neuroprotective effects, has been used as a medicinal plant in Chinese, Korean, and Japanese traditional medicine [1]. Proteins, polysaccharides, triterpenoid saponins, and flavonoids have been isolated and identified from this plant [2, 3]. Chaenomeles sinensis (Thouin) Koehne fruit (Rosaceae) has been widely used as a traditional Chinese medicine to treat throat disease. Many researchers have confirmed that this fruit have antioxidant, antitumor, anti-inflammatory, and immunoregulatory activities with the phytochemical ingredients of flavonoids, triterpenes, phenolics, carbohydrates, amino acids, and tannins [4–6]. Glycyrrhiza species (Leguminosae) have long been recognized as a medicinal plant in traditional oriental medicine, and are widely used due to their diverse pharmacological properties including vasorelaxant, anti-inflammatory, and anti-oxidant effects [7–9]. There is growing evidence for Glycyrrhizae radix, the root of Glycyrrhiza uralensis, with beneficial effects in brains [10–12].
Ischemic stroke results from a transient or permanent reduction in cerebral blood flow caused by the occlusion of a cerebral artery via an embolus or local thrombosis [13, 14]. The depletion of oxygen and ATP by the interruption of blood flow is responsible for complex pathophysiological cascades including excitotoxicity, inflammation, oxidative damage, and apoptosis of brain tissue [15]. Under ischemic conditions, insufficient supply of oxygen leads to damage to the brain via an excessive release of glutamate and activation of various cytotoxic agents, which ultimately induce neuronal injury and death [16]. Oxidative stress is believed to exacerbate the damage caused by cerebral ischemia. Cerebral ischemic injury depletes glutathione causing lipid peroxidation and consequent cell apoptosis [17]. With the remarkable advances in neuroscience research in the past decades, various strategies to counteract mechanisms involved in ischemia-reperfusion injury for stroke treatment have been tried, but the development of effective treatment for ischemic stroke is still elusive [18]. There are much efforts to develop beneficial agents from medicinal plants to achieve neuroprotection against cerebral ischemia-reperfusion injury.
Since three medicinal plants of A. elata, C. sinensis fruit, and Glycyrrhizae radix exhibit antioxidant and anti-inflammatory activities, it was hypothesized that the combination of these three plants might protect neurons against neurodegenerative diseases such as Alzheimer disease and stroke. In a previous report, an ethanolic extract of a mixture of A. elata, C. sinensis fruit, and Glycyrrhizae radix, which was named as ACG, inhibited Aβ (25-35)-induced memory impairment in mice [19]. The current study was conducted to further verify the neuroprotective effect of ACG on cerebral ischemic injury and excitotoxicity using both in vivo and in vitro studies. It was investigated the protective effect of ACG against transient focal ischemic brain damage induced by middle cerebral artery occlusion for 2 h followed by reperfusion for 24 hr (MCAO/R) in rats. In addition, the inhibitory effect of ACG on glutamate-, hydrogen peroxide (H2O2)- and hypoxia-induced neuronal death in cultured rat cortical neurons was examined to clarify possible mechanisms of its neuroprotection against ischemic brain damage.
MATERIALS AND METHODS
The young shoots of A. elata were collected at Keryong Mountain in Daejeon, Korea. The dried and sliced C. sinensis fruit and Glycyrrhizae radix were purchased from Daegu Oriental Pharm at Daegu, Korea. These plants were identified by Dr. Bangyeon Hwang, Chungbuk National University. Each 200 g of the three plants was mixed, extracted with 95% ethanol (3 L × 24 hr × 3) at room temperature and filtered through filter paper (Advantec MFS, CA, USA). The filtrate was concentrated under reduced pressure using a rotary evaporator (Heidolph Instruments GmbH & Co., Schwabach, Germany) to yield an ethanol extract (ACG, 55 g), which was then stored at room temperature until required. ACG was dissolved in dimethyl-sulfoxide (DMSO) at concentrations of 50 mg/mL and further diluted in each experimental buffer to demonstrate neuroprotective effect in cultured neurons. The final concentration of DMSO was less than 0.1%, which did not affect neuronal viability. Glutamate, H2O2, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT), and 2,3,5-triphenyltetrazolium chloride (TTC) were purchased from Sigma Chemical (St. Louis, MO, USA).
Sprague-Dawley (SD) rats (8-week-old) and pregnant SD rats for cerebral neuron culture were purchased from Daehan BioLink (Chungbuk, Korea). Animals were housed in an environmentally controlled room at 22 ± 2°C, with a relative humidity of 55 ± 5%, a 12-h light/dark cycle, and food and water ad libitum.
Before surgery, male SD rats weighing 280–300 g were fasted overnight with free access to water. Focal cerebral ischemia was induced by MCAO for 2 h followed by reperfusion for 24 hr as previously described [20]. Following evaluation of neurological score after MCAO/R, the rats were sacrificed by decapitation under anesthesia (diethyl ether) and brains were rapidly removed. The infarct and edema volumes of brain tissue were measured using TTC staining [21]. Neurological status of animals was evaluated using the Menzies method [22]. The rotarod test was performed to evaluate motor coordination of animals. All rats were trained to stay on the accelerating rotarod (Rota rod 775, IITC, Chester, MD, USA), the speed of which was increased slowly from 4 to 40 rpm within 5 min, for at least 2 min before the surgical procedures. The time they remained on the accelerating rotarod was measured after MCAO/R. The data were presented as the longest time on the rotarod of three trials [23]. ACG (10, 25, and 50 mg/kg) suspended in distilled water was administered orally three times: 0.5 hr before and 1 hr after MCAO, and 1 hr after reperfusion.
Primary cortical neuron cultures were prepared using SD rat fetuses on embryonic days 15 to 16 as previously described [21]. Neurotoxicity experiments were performed on neurons after 5-6 days in vitro. Cultured neurons were treated with 500 μM glutamate in a 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES)-buffer (incubation buffer, pH 7.4) containing 8.6 mM HEPES, 154 mM NaCl, 5.6 mM KCl, and 2.3 mM CaCl2 at 37°C for 12 hr to induce neurotoxicity. The neurons were treated with ACG 20 min prior to treatment with 500 μM glutamate [24]. To demonstrate H2O2-induced neurotoxicity, cultured neurons were treated with 100 μM H2O2 for 20 min in the incubation buffer at 37°C. After exposed to H2O2, the neurons were washed and further incubated in H2O2-free and serum-free growth medium for 15 hr (post-incubaion), and cell viability was measured. H2O2 was diluted freshly with the incubation buffer. ACG was added 20 min prior to treatment with H2O2, and were also present in the buffer during the H2O2 incubation and post-incubation [25]. To demonstrate hypoxia-induced neurotoxicity, the culture medium was removed, and neurons were washed with the incubation buffer and incubated in the same buffer containing 1.5 mM MgCl2 in a hypoxic chamber (< 2% O2 and 5% CO2, at 37°C) for 24 hr. The incubation buffer was replaced with serum-free growth medium, and the neurons were incubated for an additional 6 hr under normoxic conditions in a CO2-incubator. At the end of the incubation, cell viability was measured. ACG was applied 15 min prior to exposure to hypoxia; it also was present in the medium during hypoxic- and normoxic-incubation periods [24].
Neuronal viability was monitored by the colorimetric MTT assay as previously described [21]. Briefly, after completion of incubation with glutamate, H2O2, and hypoxia, the culture medium was replaced by a solution of MTT (0.5 mg/mL) in serum-free growth medium. After a 4 hr incubation at 37°C, this solution was removed, and the resulting blue formazan was solubilized in acid-isopropanol (0.04 N HCl in isopropanol), and the optical density was read at 570 nm using multichannel absorbance plate reader (SunriseTM, Tecan, Austria). Results were expressed as the percentage of MTT reduction, taking the absorbance of control cells as 100%.
RESULTS
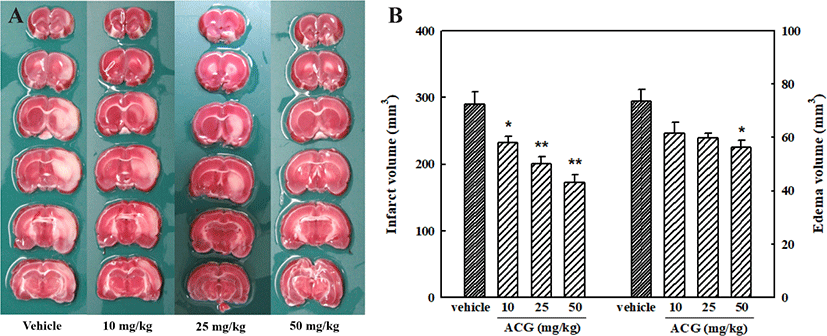
Infarction and edema are the two main pathophysiological changes observed in cases of cerebral ischemia [26]. Cellular swelling causes brain edema, which is the earliest response to ischemic injury. Representative brain slices from vehicle- and ACG-treated groups are shown in Fig. 1A. TTC-stained coronal sections in which normal brain tissue was stained deep red by a reaction of TTC with mitochondrial dehydrogenase were used to determine the volume of cerebral infarction; infarcted tissue was not stained. After MCAO/R, significant cerebral infarction was observed in the ipsilateral rat brain. Orally administered ACG (10, 25, and 50 mg/kg) reduced cerebral infarction and edema caused by MCAO/R (Fig. 1B). Rat body temperature was monitored for 6 hr after cerebral reperfusion commenced, and no significant differences were observed between the ACG and control groups (data not shown). Thus, the observed neuroprotective effect of ACG could not be attributed to hypothermia.
Degrees of neurological deficit were evaluated using the Menzies system at 24 hr after reperfusion. Sham-animals exhibited no neurological deficits (data not shown), while MCAO/R treated rats showed marked neurobehavioral deficits, such as circling movements and reduced grip strength of contralateral limbs. However, neurological scores were significantly lower in the ACG (at 50 or 100 mg/kg)-treated groups than in the vehicle-treated group (Table 1). Decreased rotarod times of MCAO/R rats were markedly recovered by ACG administration (50 or 100 mg/kg) (Table 1).
| Group | Dose (mg/kg) | Neurological score | Rotarod duration (s) |
|---|---|---|---|
| Vehicle | - | 2.5 ± 0.3 | 54.5 ± 6.8 |
| ACG | 10 | 1.8 ± 0.2 | 89.7 ± 12.7 |
| ACG | 25 | 1.2 ± 0.2** | 122.2 ± 18.3* |
| ACG | 50 | 1.2 ± 0.2** | 141.2 ± 10.5** |
After MCAO/R, neurological status and rotarod duration of the animals used in Fig. 1 were evaluated. Five categories of neurological finding were scored: 0, no apparent deficits; 1, contralateral forelimb flection; 2, decreased grip of contralateral forelimb; 3, contralateral circling of pulled by tail; 4, spontaneous contralateral circling.
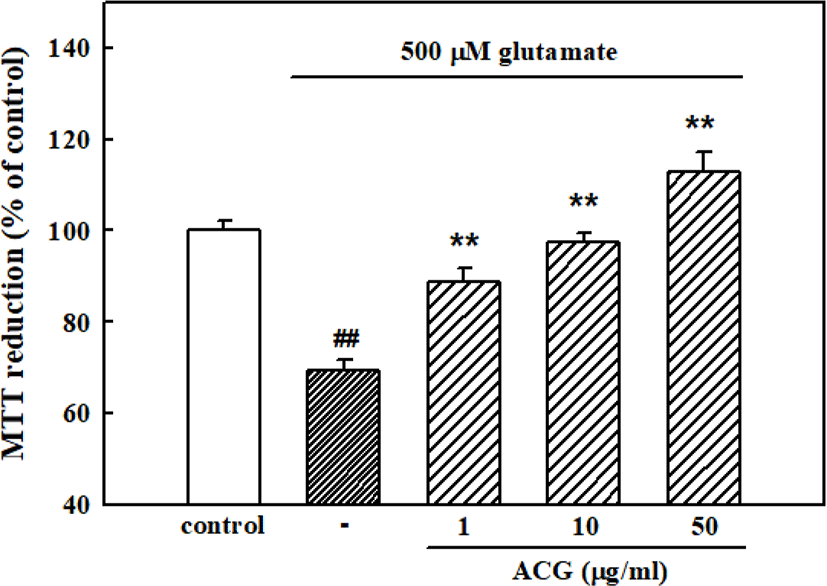
When cortical neurons were exposed to 500 μM glutamate for 12 hr, absorbance in the MTT assay was 69.3 ± 2.3% of that of the untreated control, indicating that glutamate caused neuronal death. Pretreatment of cortical neurons with ACG inhibited neuronal death induced by 500 μM glutamate showing 88.9 ± 2.7%, 97.3 ± 2.2%, and 112.8 ± 4.4% of cell viability with 1, 10, and 50 μg/mL ACG, respectively (Fig. 2).
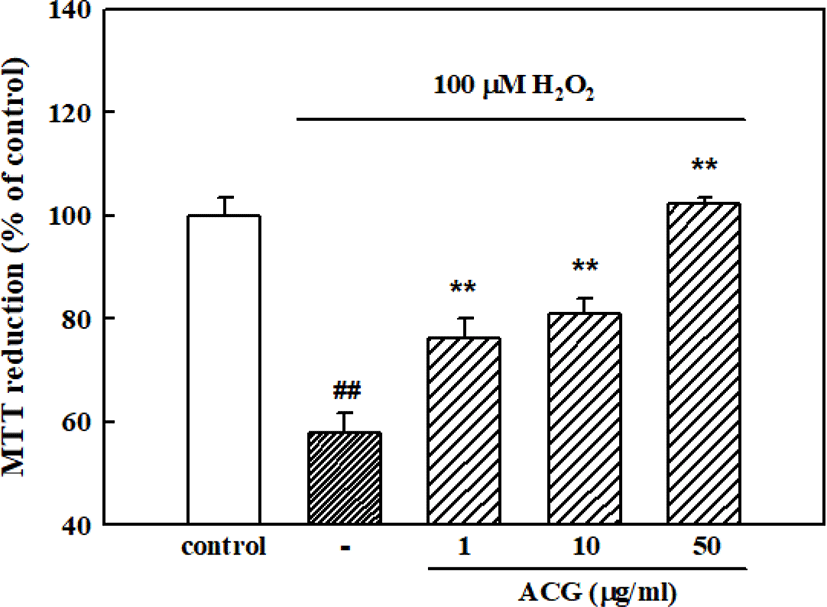
The concentration of 100 μM of H2O2 was used for determining H2O2-induced neuronal cell damage in the present experiments based on our previous result [27]. When cortical neurons were exposed to 100 μM H2O2, absorbance in the MTT assay was 57.8 ± 3.8% of that of the untreated controls, indicating that H2O2 caused neuronal cell death. Pretreatment of cortical neurons with 1, 10, and 50 μg/mL ACG reduced the neuronal death induced by 100 μM H2O2 (absorbance, 76.2 ± 3.9%, 81.0 ± 2.9%, and 102.2 ± 1.2% of control, respectively; Fig. 3).
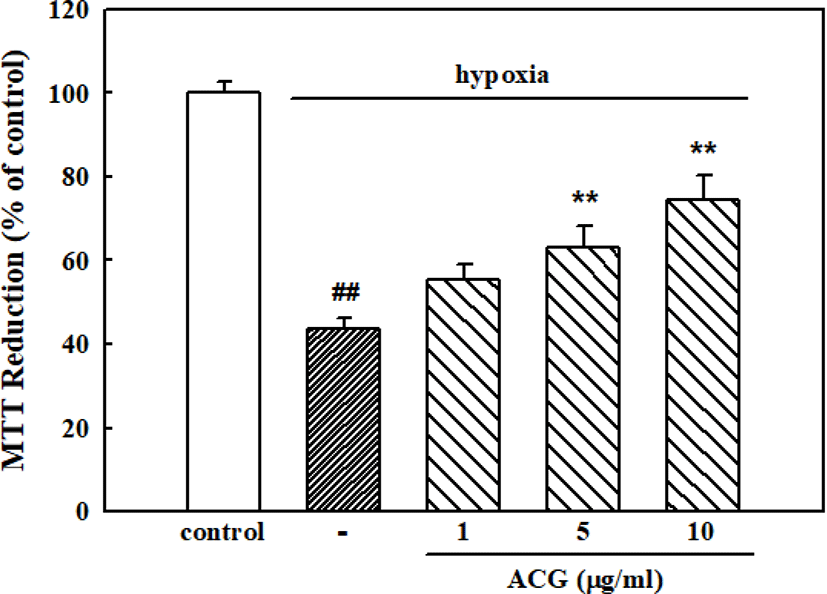
Neurons exposed to hypoxic conditions in the serum-deprived, glucose-free HEPES buffer for 24 h showed neuronal cell death, resulting in 43.8 ± 2.3% cell viability compared with the control in normoxic condition. However, ACG significantly inhibited hypoxia-induced neuronal cell death showing 63.0 ± 5.2% and 74.6 ± 5.7% of cell viability with 5 and 10 μg/mL ACG, respectively (Fig. 4).
DISCUSSION
The focal cerebral ischemia model with transient MCAO/R in experimental animals is generally accepted as the most appropriate model for human stroke [28]. It has been reported that MCAO/R model by intraluminal suture method in rats induces primary damages in the subcortical striatum and expands secondary damages into the cortical region, similarly to human stroke [29]. Neurological deficits are correlated with the increased infarct/edema size in the frontal cortex and striatum after MCAO/R in rats [26]. Thus, the MCAO/R model was used to induce ischemic injury in rat brain in the current study. MCAO/R produced brain infarction and edema and significant impairment of neurological function in rats in the present study. These changes were inhibited by ACG.
The neuronal death in such conditions as ischemia, hypoglycaemic coma, or cerebral trauma appears to be mediated at least in part by the extensive release of glutamate and its interaction with receptors [30]. Excitotoxicity through overactivation of N-methyl-d-aspartic acid (NMDA)-type glutamate receptors due to extensive release of glutamate is well established as the most important trigger of tissue damage in cerebral ischemia [31, 32]. Focal ischemia-induced infarct and edema were blocked by the NMDA receptor antagonist MK-801 in previous reports [21, 33]. Since the NMDA receptor is highly permeable to Ca2+, activation of the NMDA receptor elevates the influx of Ca2+. Excessive increase in intracellular Ca2+ concentration ([Ca2+]i) causes mitochondrial Ca2+ overload and reactive oxygen species (ROS) formation leading to lipid peroxidation and, resultantly, neuronal cell death [34, 35]. Therefore, the neuroprotection of ACG against glutamate-induced neurotoxicity in cultured cortical neurons was examined in the present study. Treatment of neurons with ACG significantly suppressed glutamate-induced neuronal cell death.
In vitro H2O2 toxicity has been used for studying the neuropathology of oxidative stress in central nervous system (CNS) disorders as a well-established model. H2O2-induced neurotoxicity in cultured neurons has been demonstrated to involve sustained elevation of [Ca2+]i, NMDA receptor modulation induced by glutamate release and ROS generation [36, 37]. H2O2 exposure causes neuronal cells to exhibit increased permeability to Na+ ions, resulting in membrane depolarization and subsequent a large influx of Ca2+ ions via voltage-dependent Ca2+ channels (VDCC) [38, 39]. H2O2 has been demonstrated to inhibit the uptake and enhance the release of glutamate, resulting in NMDA receptor overstimulation and a further increase in [Ca2+]i [37, 40]. In the present study, ACG inhibited H2O2-induced neuronal death in cultured cortical neurons. These results suggest that ACG might prevent glutamate- and H2O2-induced Ca2+ entry through VDCC- and/or NMDA-receptor-coupled channels to inhibit ROS generation and then neuronal death. Furthermore, ACG effectively protected cultured neurons exposed to hypoxic conditions in the present study. It, thus, was presumed that the neuroprotective effects of ACG might be due to the anti-oxidant effect.
The young shoots of A. elata, called dureup, are commonly eaten as a popular cuisine in Korea. A. elata containing various antioxidants, such as congmuyanosides, echinocystic acid and oleanolic acid, increases mental performance, working capacity, and endurance of movement and has shown the efficiency in traumatic brain injury, neurological diseases and myasthenia syndrome [1]. Aralia cordata (50 mg/kg) inhibited neurodegeneration by MCAO/R in rats [33]. The major compounds of C. sinensis fruit, triterpenoids and flavonoids, are well known for their antioxidant activity [4–6]. Phenolic constituents of C. sinensis fruit showed anti-neuroinflammatory, neurotrophic, and cytotoxic activities [41]. In a previous report, C. sinensis fruit suppressed Aβ (25-35)-induced memory impairment via inhibition of brain cholinesterase activity [42], although its anti-ischemic effect in animal brain has not been reported. Glycyrrhizae radix contains many licorice flavonoids including liquiritin, isoliquiritin, liquiritigenin and isoliquiritigenin [43]. Glycyrrhizae radix has been reported to have neuroprotective effects in a MCAO/R mice model at a dosage of 300 mg/kg [44]. Maximal effect of ACG was shown at the lower dosage (50 mg/kg) than the sum of maximal effect dosage of each plant. Therefore, it is suggested that the preparation of ACG might reveal synergistic effect of three medicinal plants in neuroprotection of ischemic brain damage and neurobehavioral deficits. Abundant antioxidants contained in this preparation might be responsible for the prevention of MCAO/R-induced brain damage.
In conclusion, it is evident that ACG could provide a marked protection against ischemia-induced neurobehavioral impairment. ACG also inhibited glutamate-, H2O2-, and hypoxia-induced neuronal cell death in cultured neurons. Therefore, it is suggested that the neuroprotective effect of ACG against ischemia-induced brain damage might be associated with its anti-excitotoxic and anti-oxidative activity and that ACG may be effectively applied for the prevention of neurodegeneration in stroke.