
INTRODUCTION
Alzheimer’s disease (AD) is an irreversible and progressive neurodegenerative disease with the highest prevalence worldwide. The most common early sign is memory loss of recent events [1]. Moreover, the exact cause of AD remains poorly understood. AD is a complex disease that has several pathological characterizations, including the formation of amyloid beta (Aβ) plaques, neurofibrillary tangles, and blood-brain barrier (BBB) dysfunction [2–4]. These are accompanied by neuronal and synaptic connectivity impairment, resulting in progressive neuronal cell loss [5, 6].
The BBB comprises basement membrane, pericytes, astrocytes, and endothelial cells, tightly connected by tight junctions (TJs) and adherence proteins [7]. The BBB has highly selective permeability by active transport and simultaneously protects the brain from circulating pathogens. Zlokovic et al. suggested that Aβ deposition in the AD brains may be due to disruption of homeostasis between its production and clearance from the brain [8]. In addition, Aβ can be eliminated through transport proteins across the BBB, degradation, and perivascular drainage via the vascular membrane [9].
AD is well known to have significant vascular pathophysiology, including disruption of BBB’s integrity and function [7]. For example, Aβ42 leads to increased BBB permeability and decreased the expression level of TJ protein by several mechanisms, such as neuroinflammatory responses, oxidative stress, decreased levels of annexin A1 (an anti-inflammatory messenger) and low-density lipoprotein receptor-related protein 1 (LRP1), and increased levels of receptor for advanced glycation end products (RAGE) and matrix metalloproteinase 9 (MMP9) [7, 10]. In addition, Wan et al. demonstrated that the oligomer Aβ1–42 is an important damaging factor that results in the upregulation of RAGE expression, finally inducing BBB leakage and disruption of TJ scaffold proteins in AD [11].
Electroacupuncture (EA) is a complementary alternative medicine technique widely used to treat pain, inflammation, and neurodegenerative diseases in East Asia. EA involves the delivery of an electrical current to the acupoints to stimulate the body. Several studies have demonstrated that EA treatment at acupoints such as Baekoe (GV20), Joksamni (ST36), Daechu (GV14), and Sugu (GV26) decreased infarction volume and improved neurological function by modulating BBB-related protein expression in a focal cerebral ischemia stroke model [12–14]. In addition, GV20 and Yindang (EX-HN3) combined with donepezil could improve learning and memory by regulating BBB permeability in senescence-accelerated mouse prone 8 (SAMP8) mice [15]. Acupuncture treatment at the KI3 acupoint activated cerebral neurons and functional connectivity in elderly patients with mild cognitive impairment through functional magnetic resonance imaging (fMRI) [16, 17]. In addition, KI3 stimulation regulated brain glucose metabolism in spontaneously hypertensive rats [18]. Furthermore, we demonstrated that EA treatment at KI3 acupoint regulated glucose metabolism in the transient vascular dementia animal model [19] and attenuated memory dysfunction by an increase of anti-neuroinflammation in the 5Xfamilial AD (FAD) model [20].
This study demonstrated the effects of EA treatment at KI3 on BBB integrity, alteration of TJ scaffold proteins, and inflammatory cytokine levels in peripheral blood circulation in 5X FAD mice.
MATERIALS AND METHODS
In this study, 5XFAD mice expressing human APP and PSEN1 transgenes were used as AD animal models. 5XFAD mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and bred as described previously [20]. DNA was extracted from 1 to 2 mm tail tissue for genotyping to identify pups using polymerase chain reaction analysis. Four mice per case were housed in specific pathogen-free conditions (21 ± 3°C, 50 ± 10%, 12-h light/dark cycle), with ad libitum access to water and food. Female 5XFAD mice used for this experiment were randomly divided into two groups: the 5XFAD transgenic mice (Tg) group and Tg treated with electroacupuncture (EA) at the KI3 acupoint (Tg + KI3) group. All animal care guidelines of the Korea Institute of Oriental Medicine (KIOM) were adhered to, and all experiments were approved by the Institutional Animal Care and Use Committee of KIOM.
EA treatment methods were previously described [20]. The 6.5-month-old 5XFAD mice were stimulated with EA at the Taegye (KI3) acupoint three times a week for two weeks. The KI3 acupoints were located in the medial aspect of the foot, between the medial malleolus and the Achilles tendon, based on the national standards of the acupoint location (GB12346-90) [20–22]. For EA treatment, 5XFAD mice were anesthetized using isoflurane (Hana Pharm, Hwaseong, Korea), and 8-mm long, 0.18-mm diameter acupuncture needles (Dongbang Medical, Boryeong, Korea) were inserted 2 mm deep, then an electrical stimulator (Partner-1; Daejeon, Korea) was connected, and the bilateral KI3 acupoints were stimulated. The stimulator run at an intensity of 1 mA and a frequency of 2 Hz for 15 min.
Mice were transcardially perfused with phosphate-buffered saline (PBS) for immunofluorescence staining. Whole brains were removed and kept in 4% paraformaldehyde overnight for fixation, then immersed in a 30% sucrose for osmotic dehydration at 4°C. Brain tissues were coronally cut at 30 μm thickness for immunostaining using a cryostat (Leica Microtome, Wetzlar, Germany).
The prefrontal cortex was carefully isolated and homogenized in Dulbecco’s PBS (DPBS) using a Dounce grinder for western blotting. The homogenate was separated by centrifugation at 2,000 × g for 5 min at 4°C. After centrifugation, the supernatant was removed, and the pellets resuspended in 15% (wt/vol) dextran–DPBS, then centrifuged again at 10,000 × g for 15 min. The supernatant (brain tissue-containing layer) was discarded, and the pellet (microvessel) resuspended in 1 mL DPBS, transferred to a 20-μm cell strainer, and washed with < 10 mL of DPBS. Finally, the filter was reversed, and microvessels retrieved using 10–20 mL of DPBS and centrifuged at 5,000 × g for 10 min to yield the final microvessel pellet. The supernatant was discarded and added 80 μL of ice-cold protein lysis buffer (Biosesang, Seongnam, Korea) with inhibitors (Thermo Fisher Scientific, Waltham, MA, USA).
To measure inflammatory cytokine levels in mouse serum, whole blood samples were collected from the heart before euthanasia, as previously described [23]. Whole blood was allowed to clot at room temperature, separated by centrifugation, and the supernatant was collected. The supernatant was designated as serum and transferred into a new tube for the next experiment. The inflammatory cytokine levels in the serum of mice were measured using LEGEND plex cytokine bead arrays (BioLegend, San Diego, CA, USA). Undiluted serum was used in the experiment, following the manufacturer’s instructions. High-performance flow cytometry (BD, East Rutherford, NJ, USA) was used to measure and analyze the data.
The western blotting procedure was used as previously described [20]. The homogenate (20 µg of proteins) was separated on Bolt 4%–12% Bis-Tris Plus gels (Thermo Fisher Scientific) and transferred to PVDF membranes. Membranes were incubated in 5% skim milk for blocking and then incubated in anti-aquaporin 4, occludin, and CD31 antibodies (1:1,000; Abcam, Cambridge, MA, USA) overnight at 4°C. The next day, the membranes were immersed in indicated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were developed using ChemiDoc Imaging System device (Bio-Rad) for visualization. Images were analyzed using ImageJ (version 1.46j; National Institutes of Health, Bethesda, MD, USA) for quantification.
For immunofluorescence studies, free-floating sections were blocked in bovine serum albumin at room temperature, and then immersed in the indicated primary antibodies: anti-aquaporin 4 (1:1,000; Abcam, Cambridge, MA, USA), anti-CD31 (1:1,000; Abcam), and anti-claudin 5 (1:1,000; Thermo Fisher Scientific) overnight at 4°C. Then sections were incubated in goat anti-mouse Alexa 488 (1:1,000; Invitrogen, Carlsbad, CA, USA). For visualization, images were acquired using fluorescence microscope (Olympus BX53).
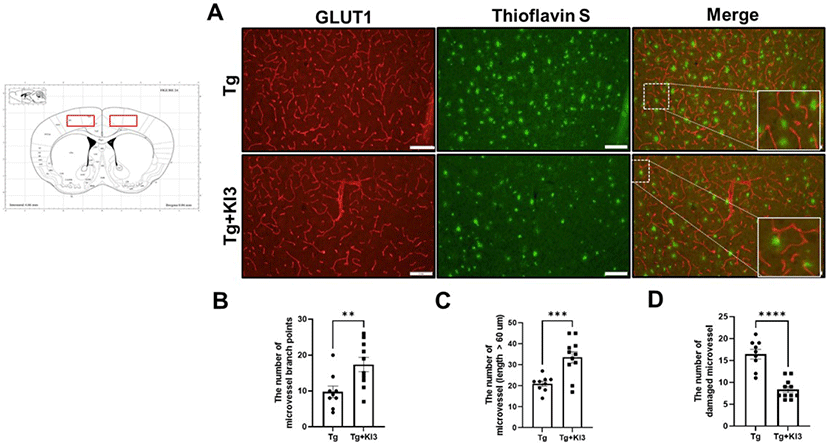
For double immunofluorescence staining, free-floating sections were blocked and incubated in glucose transporter 1 (GLUT1) (1:1,000; Abcam) antibody overnight at 4°C. Sections were immersed in matched secondary goat anti-rabbit Alexa 598 (1:1,000; Invitrogen). After antibody staining, sections were washed in PBS and stained with 1% thioflavin S for 10 min to visualize fibrillar Aβ. The number of microvessel branch points, microvessels measuring > 60 µm, and damaged microvessels (near the Aβ plaques and displayed as disconnected short length forms) were counted per section, per animal, by a researcher blinded to study data.
RESULTS
Although GLUT1 plays an important role in maintaining BBB integrity, GLUT1 deficiency in endothelial cells induces BBB breakdown in AD mice [24]. In addition, Kook et al. demonstrated that Aβ1–42 disrupts cerebral capillaries and BBB integrity in 5XFAD mice [25]. Based on these studies, we first determined whether BBB integrity was improved by EA treatment using GLUT1 immunostaining (Fig. 1A). As shown in Fig. 1B and 1C, the number of microvessel branches and microvessels (length > 60 µm) significantly increased by 1.8- and 1.6-fold in the EA treatment in the KI3 group compared with that in the non-treated Tg group in the prefrontal cortex of 5XFAD mice. Kook et al. also observed disconnected short microvessels near Aβ plaques in 5XFAD mice [25]. We also confirmed whether EA treatment ameliorates damaged microvessels near Aβ plaques by co-staining with endothelial markers (GLUT1) and Aβ1–42 plaques (Thioflavin S) and found that the number of abnormal microvessels was significantly decreased by 2-fold in the EA treatment group compared with that in the non-treated Tg group (Fig. 1C). Our results indicate that EA treatment attenuates BBB integrity in the prefrontal cortex of 5XFAD mice.
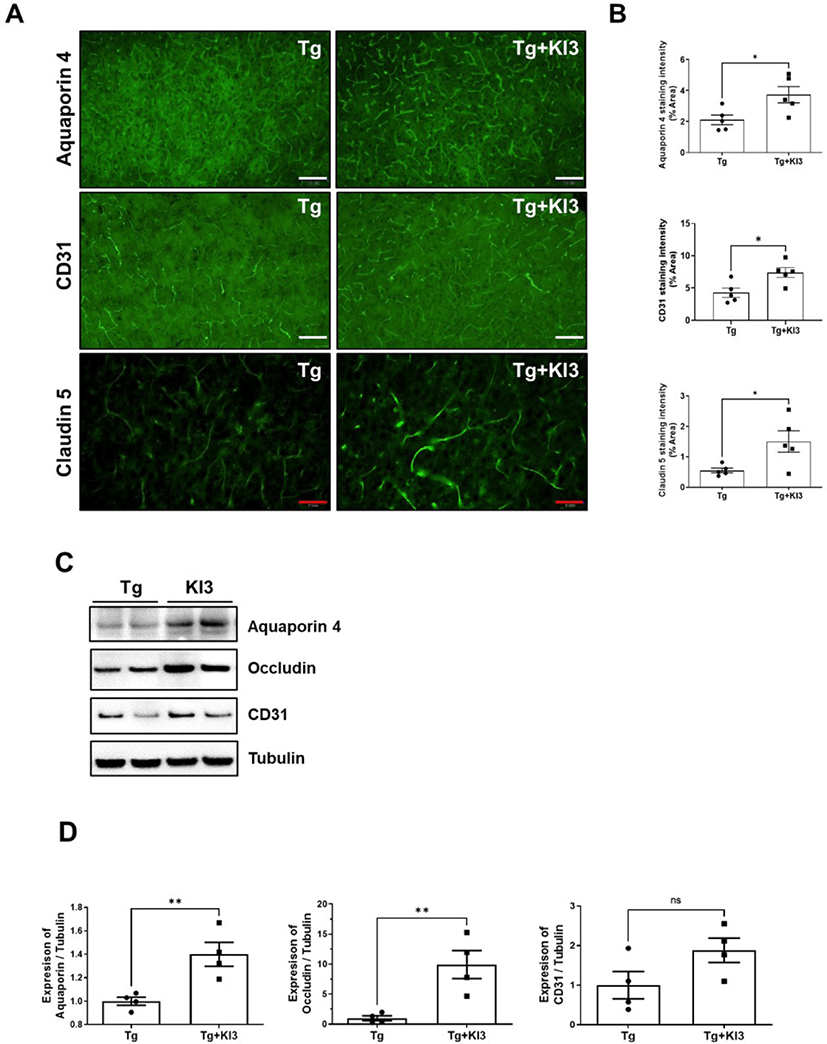
Previous reports have suggested that Aβ deposition induces several related proinflammatory and cytotoxic factors in the vasculature of the AD brain [26, 27]. Additionally, some studies have reported that Aβ deposition regulates TJ-related proteins, including occludins, claudins, and ZO, and finally disrupts BBB’s physiological function in AD patients [25, 28, 29]. Therefore, we demonstrated that EA treatment at KI3 changes BBB TJ proteins in the prefrontal cortex of 5XFAD mice through immunohistochemical staining. First, we determined the intensity of immunostaining for perivascular astrocytic end-feet marker (aquaporin 4), endothelial cell marker (CD31), integral membrane protein, and TJ strand marker (claudin 5). As shown in Fig. 2A and 2B, the levels of BBB TJ-related markers, aquaporin 4, CD31, and claudin 5, were significantly increased by 1.8-, 1.7-, and 2.7-fold in the KI3-EA treatment group compared to those in the non-treated Tg group. We also observed protein expression of BBB TJ-related markers in a microvessel isolated sample of the prefrontal cortex using western blotting. As shown in Fig. 2C and 2D, the group that underwent EA treatment showed significantly increased protein expression of aquaporin 4 and occludin (integral membrane protein) by 1.4- and 9.9-fold, respectively, compared to the non-treated Tg group in the microvessel isolated samples. In addition, the protein expression of CD31 increased by 1.9-fold compared to the non-treated Tg group , but the difference was not significant. Collectively, these data show that EA treatment at KI3 improves integrity through BBB TJ in the prefrontal cortex of 5XFAD mice.
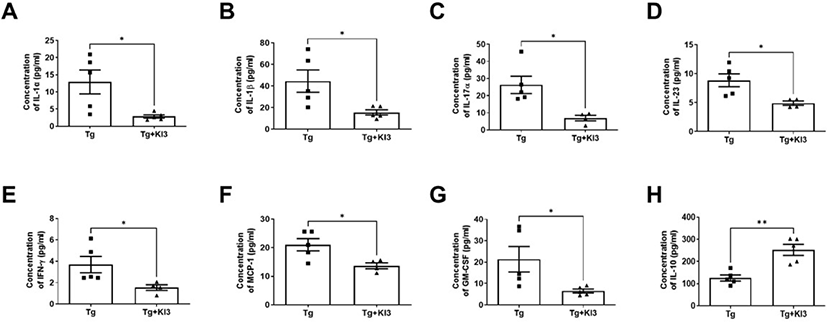
Several reports showed altered levels of several cytokines in the serum of AD mice, including increased proinflammatory cytokines such as IL-1α, IL-1β, TNF-α, and monocyte chemoattractant protein 1 (MCP-1), and decreased anti-inflammatory cytokine IL-10 [30–32]. In addition, Dubenko et al. showed that IL-17 and IL-23 levels were significantly higher in AD patients than in controls [33]. Therefore, in this study, we investigated changes in proinflammatory cytokines in the peripheral circulation of 5XFAD mice. Before euthanizing the mice, serum samples were harvested to analyze the level of inflammatory cytokines using flow cytometry (Fig. 3). The results showed that mice who underwent EA treatment at KI3 showed significantly reduced levels of proinflammatory cytokines IL-1a, IL-1β, IL-17, IL-23, IFN-r, MCP-1, and GM-CSF by 4.5-, 2.9-, 3.8-, 1.8-, 2.4-, 1.5-, and 3.3-fold, respectively, and dramatically increased IL-10 levels by 2-fold compared to non-treated Tg mice in the serum of 5XFAD mice. Therefore, these data indicate that EA treatment at KI3 regulates pro- and anti-inflammatory cytokines in the peripheral circulation of 5XFAD mice.
DISCUSSION
The present results demonstrate that EA treatment at the KI3 acupoint attenuates BBB integrity by protecting BBB TJs in the prefrontal cortex of 5XFAD mice. In addition, inflammatory cytokines were regulated by EA treatment at KI3 in the peripheral circulation of 5XFAD mice.
EA treatment has been effectively used to protect cognition via anti-inflammatory and antioxidative stress and to regulate metabolism in AD patients and mice [34–36]. In addition, EA treatment reportedly ameliorates BBB integrity in focal cerebral ischemia and the SAMP8 animal model [15, 37, 38]. A previous study demonstrated that EA treatment at the KI3 acupoint improves cognitive impairment through anti-neuroinflammatory effects in 5XFAD mice. Interestingly, we also found that microglia-mediated Aβ degradation decreased by EA treatment at KI3 [20]. Therefore, in this study, we first attempted to investigate the effects of EA treatment at KI3 on AD-associated BBB disruption, which appears to be a pathophysiological mechanism in neurodegenerative diseases, including AD and vascular dementia [39, 40]. We confirmed BBB integrity using GLUT1 immunostaining, where GLUT-1 is limited to expression in endothelial cells and lining the brain microvasculature [25, 41]. We observed that the distribution of > 60-µm long individual microvessels and microvessel branch points increased by EA treatment at KI3 (Fig. 1). In addition, previous reports have suggested that abnormal and disrupted cerebral capillaries are present near Aβ plaques in 5XFAD mice [25]. Based on the above, our data showed that EA treatment significantly decreased the number of damaged microvessels near Aβ. Thus, we concluded that EA treatment ameliorates AD-associated BBB disruption in 5XFAD mice.
Many studies have suggested the relationship between BBB dysfunction and the pathophysiological mechanisms of AD. These results show increased BBB permeability and altered structure of the BBB, including endothelial cells, astrocyte end-feet, pericytes, and basement membrane, in AD patients and mice [4, 26, 42]. Endothelial cells are the major components of the BBB and connected by TJs, and other neurovascular units contribute to BBB integrity [43]. In addition, Magaki et al. suggested decreased expression of CD31, collagen IV (COL4), and Claudin-5 and increased BBB leakage in AD patients [44]. In addition, Yao et al. demonstrated that the reduction of astrocytic laminin decreases aquaporin-4 and the level of TJ protein and disrupts BBB integrity through the regulation of pericyte differentiation [45]. Moreover, a previous in vitro study suggested that Aβ deposition decreases ZO-1 and occludin levels and disturbs the organization of claudin-5 to contribute to BBB function [42]. Based on these previous studies, we found that EA treatment at KI3 in 5XFAD mice significantly reduced the intensity of aquaporin 4-positive astrocyte end-feet, CD31-positive endothelial cells, claudin 5-positive endothelial TJs, and the protein expression of aquaporin 4, occludin, and CD31 in the prefrontal cortex compared to that in non-treated 5XFAD mice (Fig. 2). Consequently, EA treatment at KI3 decreased BBB integrity and abnormal microvessels by protecting BBB TJ proteins in 5XFAD mice.
The BBB is an important physiological barrier between the peripheral circulation and the central nervous system (CNS). Alvarez et al. reported that the levels of IL-1β and TNF-α were increased in the blood of AD patients [31] and another group demonstrated that the level of BDNF, TNFα, and IL-10 in the serum changed in early or late onset AD [32]. Additionally, Aβ deposition provides ligands to activate microglial Aβ phagocytosis, while also producing microglia-mediated proinflammatory cytokines [46]. IFN cytokines play an important role in activating microglial responses in AD and aging [47]. Yamamoto et al. indicated that IFN-γ induces microglial TNF-α, and enhance Aβ deposition through BACE1 expression and suppression of Aβ clearance in in vitro studies [48]. In addition, Steeland et al. demonstrated that TNF receptor 1-deficient APP/PS1 mice ameliorated Aβ levels and inflammatory factor expression, improved CSF-blood barrier integrity, and improved memory [49]. In addition, blocking the IL-1 cascade ameliorates brain inflammatory responses, cognition, and tau pathology, and partially reduces certain fibrillar and oligomeric forms of Aβ. In addition, Choi et al. demonstrated that normal human astrocytes express several cytokines and chemokines, including IL-6, granulocyte and granulocyte-macrophage colony stimulating factors (GM-CSF), and MCP-1/CCL2. Additionally, activated astrocytes by IL-1β and TNF-α were the newly secreted IL-1β, IL-1ra, TNF-α, and MIP-1α (CCL3) [50]. In addition, astrocyte stimulation with IL-17 increases proinflammatory cytokines and neutrophil-mobilizing cytokines and chemokines, including GM-CSF, CXCL2 (MIP-2), MMP9 [51]. Furthermore, activated microglia and astrocytes release inflammatory cytokines (CXCL8/IL-8, CCL2/MCP-1, TNF-alpha, IL-1beta/IL-1F2), leading to peripheral immune cell entry into the CNS. Leukocytes, a circulating immune cell, migrate through the BBB into the AD brain to secrete inflammatory cytokines and trigger increased BBB permeability [26]. Therefore, inflammatory cytokines are strongly related to neuroinflammation induced by glial cell activation in the CNS and peripheral nervous system (PNS). Interestingly, our previous data showed that EA treatment of KI3 significantly reduced microglia and astrocytes in the prefrontal cortex of 5XFAD mice. In addition, the number of colocalized Aβ1–42 and microglia was dramatically reduced by EA treatment in the prefrontal cortex of 5XFAD mice [20]. The present results showed significantly reduced serum levels of inflammatory cytokines (IL-1α, IL-1β, IL-17, IL-23, IFN-ɣ, MCP-1, GM-CSF, and IL-10) after EA treatment in 5XFAD mice using flow cytometry. This means that EA treatment significantly regulates inflammatory cytokines in the CNS and PNS to protect BBB integrity and BBB TJs.
In summary, our data suggest that EA treatment at KI3 improves BBB integrity and decreases abnormal microvessels by increasing BBB TJ proteins. Additionally, inflammatory cytokines were regulated by EA treatment in the peripheral circulation of 5XFAD mice. Therefore, we suggest that EA treatment at KI3 could be a potential therapeutic agent for enhancing BBB integrity in neurodegenerative diseases. However, this study has some limitations. EA treatment parameters such as frequency and intensity should be confirmed to achieve better treatment outcomes. In addition, the molecular mechanism of BBB integrity and influx of peripheral immune cells into the CNS should be investigated by EA treatment.