
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by selective degeneration of lower motor neurons in the spinal cord and brainstem and upper motor neurons in the motor cortex. The typical age of disease onset is 60 years or 50 years in familial cases. The average time from disease onset to death is 2–4 years. Most cases of ALS are sporadic, with only 5%–10% of cases being familial. The latter are associated with mutations in genes encoding superoxide dismutase (SOD)1, ataxin 2, TAR DNA-binding protein 43, and C9orf72 [1]. SOD1G93A mutant mice exhibit ALS-like symptoms and are used as an experimental disease model. ALS has a multifactorial pathogenesis that includes excitotoxicity, autoimmunity, oxidative stress, and neuroinflammation in motor neurons; however, it is unclear how these contribute to disease development [2].
Most research on ALS to date has focused on motor neuron death as a target for therapeutic strategies. In contrast, relatively little is known about the pathological mechanisms underlying the degeneration of non-neuronal cells such as microglia, astrocytes, and muscle [3–5]. ALS patients and animal models exhibit muscle paralysis and weakness as the disease progresses. Disease onset is accompanied by motor neuron death in hSOD1G93A mice [1] and impaired motor function is correlated with muscle denervation in SOD1G93A mice [6].
ALS pathology begins at the neuromuscular junction (NMJ) with terminal axonal degeneration. Morphological and pathological changes at the NMJ in hSOD1G93A mice are pre-symptomatic hallmarks of the disease [7]. This implies that NMJs plays an important role in the initiation and progression of ALS. Muscle-targeted treatments including neurotrophic factors, vascular endothelial growth factor (VEGF), and glial cell-derived neurotrophic factor (GNDF) have been shown to improve motor function and increase survival [8–10]. In addition, vesicle-associated membrane protein-associated protein B/C overexpression delayed motor impairment and neuromuscular denervation but had no effect on neuroinflammation in the spinal cord in end-stage SOD1 mutant mice [11].
To clarify the mechanistic basis for muscle degeneration and motor neuron death in ALS, the present study compared protein expression and cytokine levels in the pre-symptomatic and symptomatic stages of the disease in a mouse model. We found that proteins related to inflammation, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and cluster of differentiation (CD)11b, and to oxidative stress, such as heme oxygenase (HO)-1, ferritin, and B cell lymphoma 2-associated X protein (Bax), were upregulated in the gastrocnemius muscle of symptomatic stage hSOD1G93A mice. This was accompanied by dysregulation of metabolism and autophagy dysfunction. We also observed an upregulation of proteins related to muscle denervation atrophy in the muscle of symptomatic-stage ALS animals. These results provide insight into the molecular mechanism underlying muscle atrophy in ALS.
Materials and Methods
Hemizygous human (h)SOD1G93A transgenic (Tg) mice, which carry a mutant human SOD1 cDNA inserted randomly into the mouse genome [12], were purchased from the Jackson Laboratory (B6SJL-Tg(SOD1*G93A)1Gur) (Bar Harbor, ME, USA) and maintained as previously described [13]. Male Tg mice were identified by PCR. Animals were maintained under constant temperature (21 ± 3°C) and humidity (50% ± 10%) on a 12:12-h light/dark cycle (lights on 07:00–19:00) with free access to food and water. To clarify the mechanistic basis of muscle degeneration and motor neuron death in ALS, we compared protein expression and cytokine levels in the pre-symptomatic and symptomatic stages of the disease in a mouse model. Furthermore, we focused on the difference between asymptomatic stage at postnatal month 2 (2M Tg) and symptomatic stage at postnatal month 4 (4M Tg) in the gastrocnemius of hSOD1G93A transgenic mice and not between hSOD1G93A transgenic mice and non-Tg mice. In addition, we used only male hemizygous hSOD1G93A transgenic B6SJL mice for all experiments because the incidence of ALS is higher in men than in women.
Tg mice were grouped according to age from pre-symptomatic to symptomatic (paralysis) stages of ALS [14, 15]—i.e., pre-symptomatic at postnatal month 2 (2M Tg, 26–27 g, n = 3) and symptomatic at postnatal month 4 (4M Tg, 21–22 g, n = 3). Non-Tg (4M, 30–32 g, n = 3) mice were used as a control with B6SJL. Experiments were carried out in accordance with the animal care guidelines of the Korea Institute of Oriental Medicine.
At predetermined time points, mice were anesthetized with an intraperitoneal injection of pentobarbital (60 mg/kg) in sterile saline for gastrocnemius muscle collection [16].
The gastrocnemius muscle was dissected and homogenized in radioimmunoprecipitation buffer composed of 50 mM Tris-HCl (pH 7.4), 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, and 150 mM NaCl and a protease inhibitor cocktail (Calbiochem, San Diego, CA, USA). Lysates were centrifuged at 14,000 rpm for 15 min at 4°C and protein concentration in the supernatant was determined with the bicinchoninic acid assay kit (Pierce, Rockford, IL, USA). Samples denatured with LDS sample buffer (Thermo Fisher Scientific, Waltham, MA, USA) were separated by polyacrylamide gel electrophoresis on Bolt 4%–12% Bis-Tris Plus gels (Thermo Fisher Scientific) and transferred to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA, USA) that was blocked with 5% non-fat milk in Tris-buffered saline and then incubated with antibodies against the following proteins: TNF-α, IL-1β and -6, CD11b, HO-1, ferritin, thrombospondin (TSP)-1, prospero-related homeobox factor 1, p62, and ATG 7 (all 1:1,000; Abcam, Cambridge, MA, USA); Bax, DNA-damage-inducible (GADD)45α (all 1:1,000; Santa Cruz Biotechnology, CA, USA); glyceraldehyde 3-phosphate dehydrogenase (1:1,000, Thermo Fisher, Waltham, MA, USA); microtubule-associated protein 1A/1B light chain (LC)3B, transforming growth factor (TGF)-β, and Mothers against decapentaplegic homolog (SMAD)2/3 (all 1:1,000; Cell Signaling Technology, Danvers, MA, USA); glial fibrillary acidic protein (GFAP, 1:3,000; Agilent Technologies, Santa Clara, CA, USA); and peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α (1:1,000; Novus Biologicals, Littleton, CO, USA). The membrane was then incubated with peroxidase-conjugated secondary antibodies (Rabbit IgG: Santa Cruz Biotechnology; Mouse IgG: GeneTex) and immunoreactivity was visualized using SuperSignal West Femto Substrate Maximum Sensitivity Substrate (Thermo Fisher Scientific). A ChemiDoc imaging system (Bio-Rad) was used to detect immunoreactive bands; signal intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Data were analyzed using Prism v.5.0 software (GraphPad, La Jolla, CA, USA) and are presented as mean ± SEM. Quantitative results were evaluated by one-way analysis of variance followed by the Newman-Keuls post-hoc test for multiple comparisons. Statistical significance was set at p < 0.05.
Results
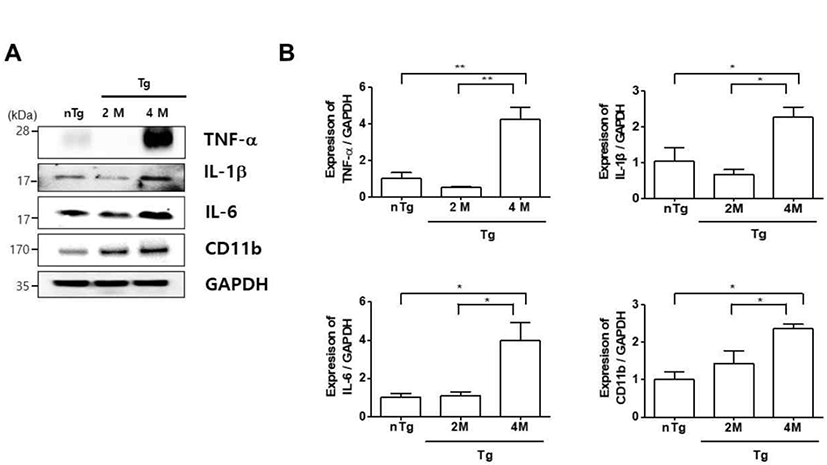
In ALS, the contractile force of motor units in the gastrocnemius muscle was reduced due to a lower number of fast-fatigable muscle fibers during ALS disease progression [17]. Therefore, we analyzed the expression of inflammation-related proteins in the gastrocnemius muscle of hSOD1G93A ALS mice, as inflammatory responses are important for maintenance of muscle homeostasis. We first examined the expression of the microglial inflammatory marker CD11b and found that it was increased in the gastrocnemius of 4M Tg 2.4 fold relative to that in nTg mice and 1.7 fold relative to that in 2M Tg mice (Fig. 1A, B). Consistent with this observation, the levels of the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 were increased 8.4, 3.4, and 3.6 fold in 4M Tg as compared to those in 2M Tg mice (Fig. 1B). These results suggest that ALS progression and NMJ dysfunction are associated with increased inflammation.
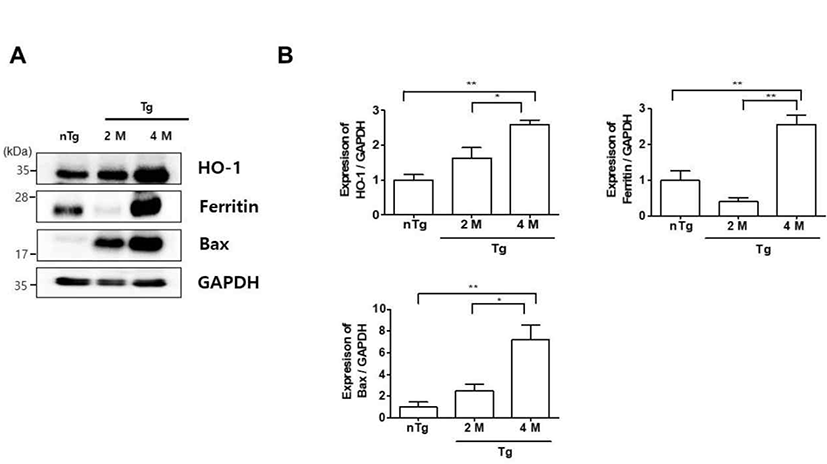
Oxidative stress is a mechanism of ALS that reflects free radical damage or abnormal free radical homeostasis [18, 19]. Inflammation and oxidative stress also contribute to the death of motor neurons in ALS [20]. To determine whether oxidative stress was associated with the observed inflammation in the gastrocnemius muscle of symptomatic hSOD1G93A mice, we examined the expression of oxidative stress-related markers by western blotting (Fig. 2). HO-1, ferritin, and Bax levels were increased 2.6, 2.6, and 7.2 fold in 4M Tg as compared to those in nTg mice and 1.6, 6.4, and 2.9 fold relative to those in 2M Tg mice. These data suggest that increased levels of oxidative stress-related proteins in the symptomatic stage trigger the expression of inflammatory proteins, resulting in muscle dysfunction in ALS.
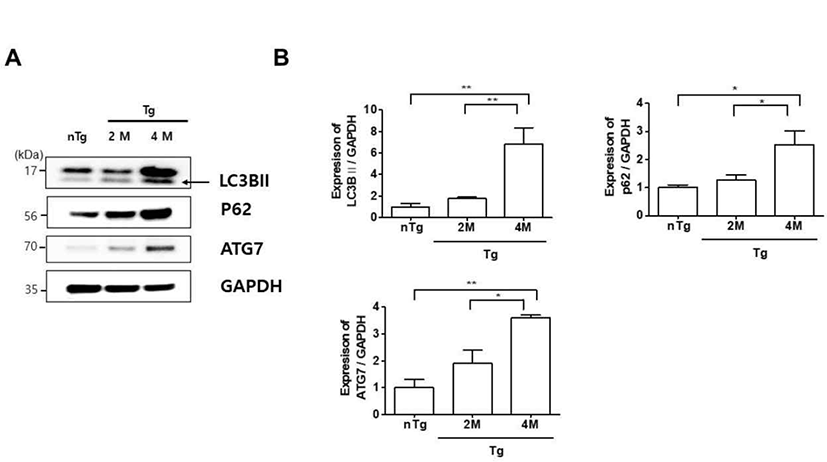
Autophagy plays a housekeeping role in cells by removing misfolded or aggregated proteins, clearing damaged organelles, and eliminating intracellular pathogens. Mutations in ALS-associated genes such as SOD1, Dynactin, and optineurin have been linked to autophagy dysfunction in mice and zebrafish [21–23]. We therefore investigated the expression of autophagy-related proteins LC3BII, p62 and ATG7 in hSOD1G93A ALS mice. The levels of these proteins were upregulated 6.8, 2.5, and 3.6 fold, respectively, in the gastrocnemius of 4M Tg as compared to those in nTg mice (Fig. 3A, B) and significantly increased by 3.8, 2, and 1.9 fold compared to those in 2M Tg mice. These data were similar to those of Olivan et al. who showed that autophagic markers including Beclin1, LC3B, and p62 were increased and suggested an enhancement of autophagic flux in skeletal muscle of hSOD1G93A mice along with disease progression [24]. Thus, autophagy dysfunction is associated with muscle degeneration in ALS.
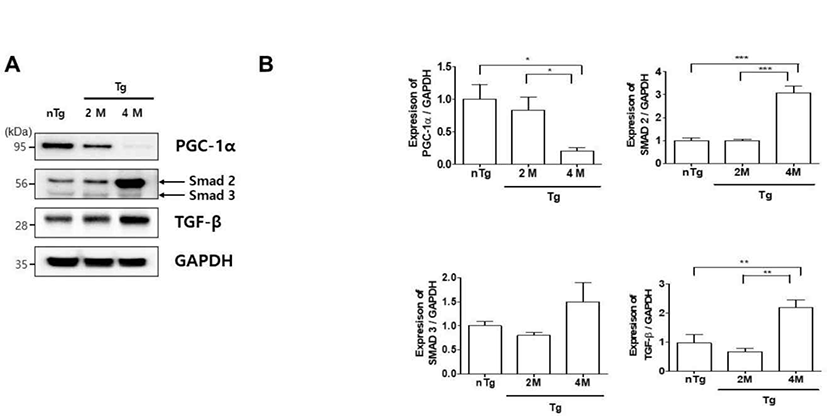
ALS patients exhibit defects in energy metabolism related to the muscle, including weight loss, hypermetabolism, and hyperlipidemia [25]. To evaluate metabolic changes in our ALS model, we compared the expression of PGC-1α, SMAD 2/3, and TGF-β in the gastrocnemius muscle of the various groups (Fig. 4A). PGC-1α was downregulated 5 fold in 4M Tg as compared to levels in nTg mice and was 4.2 fold lower in 4M Tg than in 2M Tg mice. In addition, SMAD 2, and TGF-β expression was 3 and 2.3 fold higher in 4M Tg than in nTg mice. Interestingly, SMAD 2, and TGF-β expression was significantly increased by 3 and 3.3 fold compared to that in 2M Tg mice. Thus, muscle paralysis in symptomatic ALS mice may be caused by dysregulation of metabolism and consequent inhibition of ATP synthesis.
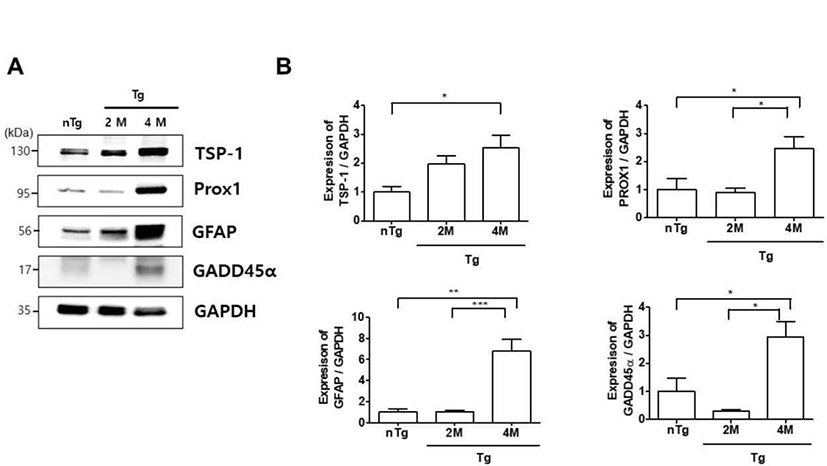
Muscle denervation leads to skeletal muscle atrophy via GADD45α and histone deacetylase activities [26]. We investigated whether changes in the expression of muscle denervation atrophy-related proteins in the gastrocnemius muscle are associated with ALS progression. TSP-1, Prospero-related homeobox (Prox)1, GFAP, and GADD45α levels were increased 2.5, 2.5, 6.8, and 2.9 fold, respectively, in 4M Tg as compared to those in nTg mice, and Prox1, GFAP, and GADD45α expression was significantly increased by 2.7, 6.6, and 9.8 fold, respectively, compared to levels in 2M hSOD1G93A mice (Fig. 5). These results indicate that upregulation of muscle denervation atrophy-related proteins leads to dysfunction of the NMJ and loss of muscle innervation by motor neurons.
Discussion
ALS is a progressive neurodegenerative disorder characterized by muscular paralysis and motor neuron death. In the CNS, neuroinflammation and oxidative stress involving microglia and astrocytes contribute to motor neuron death. However, it is unclear whether the same processes occur in muscle. To address this issue, we analyzed the expression of proteins associated with inflammation, oxidative stress, autophagy, and atrophy in the gastrocnemius muscle of hSOD1G93A mice, which served as an ALS model. At the symptomatic stage, upregulation of inflammation and oxidative stress-related proteins was linked to dysfunction of energy metabolism in muscle, associated with muscle denervation-induced atrophy. These results suggest that therapeutic strategies for ALS treatment should target the muscle at the time of disease onset in addition to motor neurons.
Following spinal motor neuron death in ALS, muscles become paralyzed, which is accompanied by inflammation and metabolic dysregulation [27]. A recent study reported an increase in inflammation in skeletal muscle near NMJs in SOD1G93A rats [28]. Consistent with these observations, we found that CD11b, IL-1b and -6, IL-6, and TNF-α levels were increased in the gastrocnemius muscle at the symptomatic stage of hSOD1G93A mice (Fig. 1). The neuroinflammation-related proteins GFAP, ionized calcium binding adaptor molecule 1, and TNF-α and related signaling molecules were previously shown to be upregulated in the spinal cord of symptomatic stage hSOD1G93A mice [13]. These results suggest that an anti-inflammatory mechanism is critical for delaying ALS progression.
Autophagy is implicated in various neurodegenerative disorders as a process that ensures the clearance of intracellular aggregates and neuronal survival [29]. In this study, we found that the oxidative stress-related proteins Bax and especially HO-1 and ferritin were highly expressed in the gastrocnemius muscle of hSOD1G93A mice at the symptomatic stage as compared to levels in non-Tg mice (Fig. 2). This was accompanied by upregulation of autophagy-lysosome pathway components (Fig. 3). Thus, increased oxidative stress and autophagy-lysosome functioning could lead to metabolic dysregulation in muscle in ALS.
ALS patients exhibit hypermetabolism, body weight loss, and abnormal lipid metabolism [30, 31]. Mattson et al. was also shown that a high-fat diet delayed disease onset, reduced weight loss, and increased survival in SOD1G93A mice [32]. However, little is known about the specific metabolic changes in motor neurons. PGC-1α regulates mitochondrial biogenesis and metabolism and is enriched in skeletal muscle [33]. Muscle atrophy in humans and aged mice resulted in a decrease in PGC-1α levels [34]. TGF-β/Smad signaling involves the direct phosphorylation of Smad2/3 by TGF-β receptor I kinase. ALS patients show elevated levels of TGF-β1 in the serum and plasma, and TGF-β1 and SMAD 2/3 expression in the muscle is increased in ALS and is considered as a disease biomarker [35]. In the present study, we observed metabolic dysregulation in the gastrocnemius muscle of hSOD1G93A Tg mice at the symptomatic stage of the disease, as evidenced by a decrease in PGC-1α and increase in SMAD 2 and TGF-β; however, Smad 3 levels did not induce disease progression (Fig. 4). Si et al. have demonstrated that Smad2 mRNA in ALS significantly increased at the 60 d post-natal stage in the beginning, which successively progressed toward the end-stage as compared with the disease controls. However, Smad3 mRNA did not induce progressive disease development in the G93A mouse. Kurisaki et al. have demonstrated that phosphorylation of Smad 3 is regulated by importin beta in the cytoplasm, while Smad 2 is imported into the nucleus by phosphorylation. Thus, the activation of Smad 2 and 3 follows two different cellular mechanisms [36]. This suggests that the activation of Smad 2 and 3 mediates different roles in muscle atrophy during the gastrocnemius of hSOD1G93A mice.
We speculate that mitochondrial dysfunction causes abnormalities in metabolism and contributes to muscle denervation and atrophy in ALS. Therefore, prophylactic treatment and presymptomatic management of diet are important for preventing or delaying ALS onset.
Muscle atrophy involves GADD45α, Prox1, and TSP-1 proteins [37]. An increase in GADD45α expression can cause muscle denervation and immobilization. Prox1 is enriched in mouse skeletal muscle and regulates the conversion of slow to fast skeletal muscle fiber function [38]. This study showed that TSP-1, Prox1, GADD45α levels were increased in 4M Tg as compared to those in nTg mice and, Prox1 and GADD45α expression was significantly increased compared to levels in 2M hSOD1G93A mice (Fig. 5). These results indicate that upregulation of muscle denervation atrophy-related proteins leads to dysfunction of the NMJ and loss of muscle innervation by motor neurons.
There are several outstanding questions that must be addressed in future studies. Firstly, the role of muscle dysfunction-related proteins in NMJ or motor neuron degradation in ALS requires clarification. Based on the important role of muscles in ALS disease onset, some papers reported that the therapeutic benefit of muscle-targeted treatments including neurotrophic factors, VEGF, and GDNF improves motor function and increases survival [8–10, 39, 40]. Furthermore, Kim et al. reported that VAMP/synaptobrevin-associated protein B (VAPB) overexpression delayed motor impairment and neuromuscular denervation in the muscle but had no effect on neuroinflammation in the spinal cord in end-stage of SOD1 mice [11]. Therefore, further investigations of the diverse molecular mechanisms underlying the relationship between motor neuron death and muscle atrophy mediated by NMJ function are required to identify effective therapy for ALS patients.
Mitochondrial dynamics in the gastrocnemius muscle must be evaluated in order to confirm the dysregulation of energy metabolism in hSOD1G93A mice at the symptomatic stage. Muscle produces and releases myokines such as IL-6, -15, and -8 and brain-derived neurotrophic factor into the circulation [41]; the relationship between these factors and the death of motor neurons in ALS must be further investigated. This experiment was conducted with three mice per group according to the animal ethics law, and significant results were obtained. However, in future studies on the difference between gender and on drug development based on muscle-target for ALS, we will increase the number of mice per group to minimize the variation between groups.
In summary, we found that inflammation and oxidative stress-related proteins were upregulated in the gastrocnemius muscle at the symptomatic stage of ALS in a male mouse model. This was accompanied by increased autophagy, metabolic dysfunction, and muscle denervation and atrophy. These findings suggest that in ALS, abnormal regulation of proteins that modulate muscle function leads to muscle paralysis. Our findings provide insight into the mechanisms underlying muscle atrophy in ALS as well as new potential therapeutic targets for disease treatment.