Introduction
20세기 후반부터 분자유전학이 발전하면서 유전자 분석기 술과 유전적 표지인자를 이용하여 양적형질에 관여하는 후 보유전자 탐색 연구가 가능하게 되었다. 다중 마커를 활용한 마커 도움 선발 연구를 진행하였으며, 2003년 인간게놈프 로젝트가 완료된 이후 시퀀싱 테크놀로지가 급격히 발전하 고, 가축에서는 2014년 닭의 표준유전체지도가 완성되며, 이후 소, 돼지, 말의 표준유전체지도가 작성되면서 각 축종 에 따라 전장유전체서열 기반의 단일염기다형성 정보가 포 함된 DNA 칩 제작이 가능해졌다. 현재는 전장유전체염기서 열을 기반의 단일염기다형성 정보가 포함된 상용칩을 활용 한 형질 예측 연구가 활발히 진행되고 있다. 이러한 발전은 유전체 분야와 육종의 융합이 가능한 새로운 패러다임을 가 져왔다. 유전체정보 기반의 유전체 예측은 크게 두 가지 방 법에 따라 구분된다. 마커도움선발이 확장된 방법의 전장유 전체연관분석은 단일염기서열다형성 정보와 형질과의 회귀 분석 또는 선형혼합모형을 기본으로 하여 단일마커와 형질 과의 연관성 분석으로 유의 수준 이하의 원인변이 마커 선정 및 선정된 마커의 기능 및 생물학적 기작 연구를 진행한다. 이 방법은 복합 형질의 경우, 유의 수준 이하의 마커만을 고 려하기 때문에, 소수의 양적형질좌위만을 설명하게 되어 해 당 형질의 유전적 효과를 모두 설명할 수 없는 문제를 야기 한다. 반면, 동·식물에서 유전체 선발이라 불리는 유전체 예 측은 마커 패널을 기반으로 연관불평형 구조 안에 하나의 마 커가 존재하게 되면 그에 따른 마커 효과를 모두 반영하여 형질에 관여하는 유전 분산을 설명해줄 수 있을 뿐만 아니 라, 전장유전체연관분석에서 범할 수 있는 다중보정 문제를 고려할 필요가 없다는 점이 특징이다 [1].
이러한 유전체 예측은 사람의 경우 질병 유발 유전인자 발 굴, 질병의 예후 및 약물반응 등 개인 맞춤형 치료를 목적으 로 연구가 진행되고 있는 반면, 동식물 분야에서는 육종 프 로그램에 활용하여 보다 정확한 육종가 추정을 통한 씨가축 선발, 경제형질 연관 원인변이 발굴에 이용되고 있다. 유전 체 예측은 유전력이 낮은 형질이나 측정하기 어려운 형질에 있어서도 가능해졌으며, 세대간격을 줄일 수 있어서 후대검 정보다 유전적 개량량을 높일 수 있고, 효율적인 비용으로 상업적으로 이용이 가능해졌다[2]. 그렇지만, 많은 연구 결 과에서 기술된 바와 같이 유전체 선발 또는 예측을 위해서 참조집단의 크기 및 형질의 유전력, 마커 간 연관불평형 등 의 집단의 유전적 구조를 파악하는 것이 우선적으로 고려되 어야 한다. 최근 양적형질좌위 및 전장유전체연관분석과 같 이 형질에 영향을 미치는 원인유전자군 및 변이를 활용한 유 전체 예측모델이 개발, 적용되면서 일부 선두 그룹에서는 다 양한 유전체정보를 기반으로 한 유전체 예측 모형이 개발되 고 있다. 이러한 연구결과는 유전체 예측에 사용되는 단일염 기서열다형성이라는 단편적인 유전체자료뿐만 아니라 고도 화된 유전체정보가 유전체 예측에 직접적으로 활용 가능하 다는 것을 시사하며, 향후 국내 유전육종 연구의 방향 및 범 위를 설정할 수 있게 된다.
본 총설에서는 유전체선발, 전장유전체연관분석 등의 유전 체예측법을 기반으로 다양한 유전체 정보가 결합되어 유전 체 예측에 활용될 수 있는 방법과 가능성 그리고 향후 연구 방향에 대하여 최근 연구 동향을 중심으로 고찰할 것이다.
Materials and Methods
전장유전체연관분석을 포함한 유전체 예측은 일반적으로 표준유전체지도로부터 절대적인 유전체상의 위치가 정해지 고, 해당 좌위에 집단 혹은 품종별로 다형성을 가지고 있는 단일염기서열다형성 마커로 제작된 상용칩을 사용하게 된 다. 최근 시퀀싱 테크놀로지가 발전하면서 상용칩에 국한되 어 연구되었던 유전체 예측법이 단일염기서열다형성 이외에 다양한 오믹스 자료를 접목시켜 유전체 예측의 정확도 향상 에 기여하는 유전적 요소들을 추가 분석하고 있다. 특히, 염 기서열 해독을 위한 제3세대 시퀀싱 장비인 옥스포드 나노 포어 및 PacBio의 single molecule, real-time (SMRT) 기술은 2세대 염기서열 해독 장비에서 이루어졌던 PCR 증 폭과정이 생략되고, 1개 분자에 대하여 염기서열 해독이 진 행된다는 것이 가장 큰 특징이다. 이를 통해 1,000달러 시 퀀싱이 가능한 시대가 도래하였다. 차세대 염기서열해독 비 용이 급감하고, 상용칩보다 많은 정보를 보유하고 있는 전장 유전체를 포함한 다양한 유전체정보를 유전체예측에 활용하 기 시작하였다. 소의 경우, 칩에 포함된 마커 수에 따라 7천 여 개, 5만개, 7십만 개 등의 다양한 상용칩이 존재하며, 대 다수의 유전체 예측 연구에 활용되었다. 상용칩을 기반으로 한 유전체예측 연구가 진행되면서 마커의 밀도를 증가시키 면 유전체예측의 정확도가 증가될 것이라 예상하였지만, 실 제로는 증가된 마커 수에 비해 정확도 향상 효과는 미미하였 다. 전장유전체염기서열자료를 시뮬레이션 하여 유전체 예 측을 진행한 결과 정확도가 약 5~10% 증가[3]하였으나, 실제 자료를 분석한 결과는 정확도에 크게 영향을 주지 않았 다[4]. 하지만, 칩을 구성하는 단일염기서열다형성은 각 품 종별 유전자형 빈도수가 높은 마커를 중심으로 제작되었기 때문에, 집단 내에 형질에 영향을 주는 원인변이가 부재하 고, 칩 내의 마커와 원인변이 사이의 연관불평형 정도를 고 려하지 않아도 되기 때문에 차세대염기서열을 활용한 유전 체예측 연구는 유전체예측이 최적화될 수 있도록 여전히 다 각적인 연구들이 진행되고 있다. 전장유전체염기서열을 활 용하여 전장유전체연관분석 수행 후, 유의한 마커군을 선 발하여 유전체예측에 활용하여 분석시간을 단축시킬 수 있 다. 뿐만 아니라, 홀스타인-프레시안 집단의 전장유전체서 열에 존재하는 36,916,855개의 변이에서 단일염기서열다 형성의 특성(이형접합성, 유전자형 타입, 연관불평형 내 존 재하는 변이 제거)을 활용하여 변이들을 제거하면서 자료의 부분집합(N=106)을 선택하기 위해 Bayesian Stochastic search variable selection (BSSVS)를 활용하여 유전체육 종가를 추정한 결과, 전체 변이정보를 사용하는 것 보다 연 관불평형 기반의 변이정보를 선택하여 자료를 구성한 것이 약 1.4% 정확도가 향상된 것을 보고 하였다 [5].
염기서열 내에 존재하는 단일염기서열다형성은 구조와 기 능적인 측면으로 구분할 수 있다. 게놈 상의 유전자 위치와 연계하여 단백질 기능을 변화시킬 수 있는 여부에 따라 단 일염기서열다형성은 coding SNPs, non-coding SNPs, regulatory SNPs 등으로 분류될 수 있다.
이들과 형질에 영향을 주는 대사회로에 관련된 유전자에 존재하는 SNPs들로 구성할 수 있다. 단일염기서열다형성과 관련된 유전자와 생물학적 대사회로를 활용하기 위해서는 BioCarta(www.biocarta.com), KEGG(Kyoto Encyclopedia of Genes and Genomes, www.genome.jp/kegg), Reactome(www.reactome.org) 등의 데이터베이스를 활 용하여 형질에 연관된 유전자군을 정의하여 유전체 예측을 시도할 수 있다. 뿐만 아니라, 유전체 기능 분석에 가장 대 표적인 Gene Ontology는 다양한 계층적 구조에서 유전자 와 유전자 산물의 기능적 특징을 정리한 데이터베이스로 유 전자 기능 주석달기 결과를 활용하여 손쉽게 형질과 연관된 유전자군을 추출할 수 있다. 유전자의 생물학적 기능이 고 려된 게놈 상의 유전체 영역에서 공통 조상으로부터 개체별 로 공유하고 있는 유전체 영역 내에 존재하는 변이 정보들을 이용하여 유전자의 기능과 구조 기반의 유전체 예측을 수행 할 수 있다. 대표적인 예로, Linkage Disequilibrium Adjusted Kinships (LDAK) 분석법은 게놈을 위치에 따라 분 류한 마커 정보를 활용하여 복합형질에 대한 원인변이의 효 과를 추정한다 [6]. Yang 등은 유전력(h2) 추정에 있어 혈 연계수를 균등하게 배분하는 것을 제안 하였으나[7], 이 경 우에는 연관불평형에 의해 발생하는 평균 바이어스를 고려 해야 하며, 변이에 대한 MAF 분포 등 사전 정보를 확보해야 한다. 이 분석법은 SNP과 그 주변에 있는 SNPs (tagging SNPs)에 대하여 가중치를 다르게 주어서 혈연관계행렬을 구성하여 유전체 예측을 진행한다. Genomic Best Linear Unbiased Prediction (GBLUP)에서 확장된 Genomic Feature Best Linear Unbiased Prediction (GFBLUP) 또한 유사한 원리로 제안된 모델이다[8]. 특정 형질에 관여 하는 유전체 영역 내 존재하는 변이들은 예측 모델에서 가 중치를 주는 방법으로 생물학적 정보가 추가되어 형질 예측 에 좀 더 정확한 추정이 가능하다는 것이다. 더 나아가 유전 체 예측을 위하여 반수체 정보도 활용될 수 있다. 반수체 분 석은 단일염기서열다형성 분석에서 밝혀지지 않은 대립 유 전자의 효과를 설명하는 유용한 도구가 될 수 있다[9]. 반수 체는 연관불평형 내에서 형질에 연관된 단일염기서열다형성 의 조합으로 양적형질좌위와 같은 기능을 가지고 있다. 집단 내에서 공유하는 반수체 길이가 길수록 분석에 유효한 정보 로 활용된다. 특히, 반수체 블록을 구성하는 방법 또한 멀티 오믹스 정보가 활용된다. 예전에는 두 마커 간의 연관불평형 측정값인 D 값의 일정 수준 이상인 마커들로 반수체를 구성 하였지만, 최근 들어 대사회로, 유전자 발현, 메틸레이션, 선 발신호 등의 유전자의 기능 정보를 추가하여 반수체를 구성 하게 된다 [9]. 홀스타인 집단의 유단백 형질의 유전체 예측 연구에서 SNP 패널 데이터로 구축된 모집단의 반수체형을 활용하여 유전체 예측을 진행할 경우 3.1%의 정확도가 향 상되는 것을 확인할 수 있었다[10].
차세대염기서열기술의 발전은 멀티 오믹스 자료를 통합하 고, 해석하기 위한 대형 프로젝트를 가능하게 하였다. 시스 템 생물학을 기반으로 멀티 오믹스 자료를 통합하여 유전체 예측에 응용하기 위한 Functional Annotation of Animal Genomes (FAANG) 컨소시움이 2015년 착수되었다[11]. 동물의 게놈에 대한 기능 주석달기를 진행하여 주요 원인이 되는 유전 정보를 찾고자 하는 시도이며, 주로 후성유전체학 적 접근을 통해 그 동안 유전체 예측에 있어 환경 효과로 처 리하였던 부분을 정밀히 연구하여 DNA에서 유전적 효과로 설명되지 않는 표현형적 특성을 연구하여 이를 유전체 예측 에 활용하고자 시도하고 있다. 후성유전체학의 하나의 사례 인 각인유전자는 유전자의 발현이 부, 모에 따라 달라지는 현상으로 멘델 유전에 따르지 않기 때문에 과거에는 이러한 현상을 환경효과, 또는 오차로 처리되어 분석에서 제외되었 다. 하지만 시퀀싱 기술이 발전하면서 각인유전자 및 유전자 내에 존재하는 변이와 경제형질 연관성 분석[12], 각인 효 과에 의한 부, 모 계통의 독립적 개량 프로그램 개발을 위한 유전체 예측 모델이 개발되기도 하였다[13]. 그 밖에, 전사 체, 단백질체, 메타게놈, 마이크로비옴 등이 함께 멀티 오믹 스 자료로 활용되어 유전체 예측에 활용될 수 있다[14].
상용칩에 국한되어 진행된 유전체 예측은 최근 몇 년 사 이 급격하게 발달한 시퀀싱 테크놀로지로 인해 High- Throughput Omics (HTO) 시대에 도래하였다. 이러한 HTO는 유전체, 후성유전체, 전사체, 단백체, 대사체, 메타 게놈 등 표현형과 직·간접적으로 연결된 생물학적 자료를 의 미하며, 동물의 생산성, 질병저항성, 복지 등 복합형질을 다 양한 생물학적 자료를 연계시켜 가축 개량 및 유전체 예측 에 활용하고자 하는 시도를 하고 있다. 가축 개량 분야에서 는 ‘시스템 유전학’ 또는 ‘시스템 유전체학’이라 불리 우며, 게놈/엑솜, 유전자 발현 칩, 차세대염기서열 해독과 같 은 ‘오믹스’ 자료를 통합하여 분석하는 분야로 인식된다. 멀티 오믹스 자료는 형질과 연관된 유전자 및 관련 변이들의 탐색 정도에 따라 제1종 오류를 감소시키는 역할을 하여 정 확한 양적형질좌위를 정의할 수 있게 된다. 차세대염기서열 자료는 상용칩의 단일염기서열다형성 정보를 포함할 뿐만 아니라, 삽입결손, 복제수변이, 초위성체마커 등 DNA에 존 재하는 유전체 변이정보를 확보할 수 있다. 예를 들어, 사람 에서 진행된 Encyclopedia of DNA Elements (ENCODE) 프로젝트에서 밝혀진 것처럼 유전체 내의 많은 기능적 모티 프가 코딩영역 외부에 존재하여, 주어진 특성에 대한 잠재적 인 인과 관계를 정확하게 나타내지 못한다[15]. 단백질 코 딩 유전자에 예상되는 효과에 따라 이들 변이형을 분류하면 유전자 기능에 가장 영향을 미치는 변이형이 밝혀 질 수 있 다[16]. 대사 및 신경 회로, 유전자 조절 네트워크, 단백질 상호작용 네트워크 등은 유전자 간의 상호작용을 정리하여 전사조절인자, microRNA 등과 연결시킬 수 있는 정보를 제 공해준다. microRNA, 전사조절인자를 포함하여 종 특이적 유전자 정보는 세대 간 유지된 양적형질좌위의 반영이 가능 하므로 유전자 기능 해석에 활용될 수 있다 [17]. 유전체 예 측에 활용 가능한 유전체 자료는 Figure 1에 설명하였다.
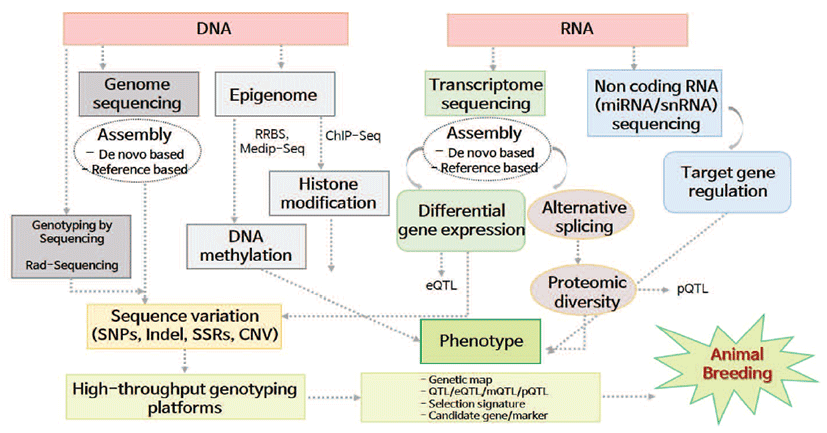
멀티 오믹스 정보를 활용한 유전체 예측은 표현형에 직간 접적으로 영향을 주는 게놈 정보를 보다 효율적으로 이용함 으로써 정확도를 향상시키고자 함이다. 가장 대표적이며 대 형 프로젝트인 1,000 bulls genome project는 호주를 중 심으로 북미, 유럽 등의 씨수소 1,000두에 대한 전장유전체 서열을 생산하여 전장유전체연관분석, 유전체 예측 연구를 통해 유전체정보 활용의 최적화 및 형질 연관 원인 유전자 탐색을 목표로 연구가 진행되고 있다. Jersey, Fleckvieh, Holstein-Fresian 집단에서 생산된 234두의 전장유전체 염기서열 자료를 활용하여 배아 손실, 모질, 연골형성장애에 관련된 원인변이를 찾고, 대용량 SNP 칩 자료에서 차세대 염기서열자료의 활용 가능성 탐색의 일환으로 결측치 추정 의 효율적 분석방안에 대한 연구도 함께 진행하였다 [18]. 최근에는 육량, 육질, 유생산 형질을 넘어 새로운 표현형 추 정을 위해 유전체 예측을 시도하기도 한다. 호주 젖소 집단 에서는 기후변화를 대비하여 고온 스트레스에 적응하는 개 체 선발을 위해 2003년부터 2013년까지 수집된 일일 온 도, 습도 자료와 함께 약 36만두의 홀스타인 집단과 7만 6 천두의 져지 집단에서 온습도 지수에 대한 유량, 유단백, 유 지방 형질에 대한 유전체 예측을 진행하였다[19]. 호주 앵 거스 집단(N=747)에서 지구 온난화에 영향을 주는 온실 가스 중, 메탄 가스 배출량에 대한 유전력을 추정한 결과, 0.19~0.29로 확인되었다. 메탄 표현형 자료를 활용하여 유 전체 예측을 분석한 결과 메탄 방출량에 대한 유전체 육종가 의 정확도는 약 0.3으로 분석되었으며, 유전체 육종가를 기 반으로 개체 선발 시에 10년 동안 메탄가스 방출량이 5% 감소할 것이라 추정하였다 [20]. 또한, 호주 앵거스 집단의 사료효율에 대한 유전체 예측을 위해 사료효율이 높은 집단 과 낮은 집단에서 태어난 자손들에 대한 사료효율을 측정하 고, 도축 후 근육, 간, 지방, 뇌하수체, 십이지장 조직에 대한 RNA-Seq 자료를 생산하여 유전자 발현값을 유전체 예측 모델에 적용하여 유전체 육종가를 비교하기도 하였다 [21]. GFBLUP 모델을 활용하여 홀스타인 집단에서 유선에 감염 을 시킨 후, 시간대 별로 간의 생검시료를 확보하여 RNASeq 자료를 분석하였으며, 차등발현 유전자내에 존재하는 SNP 에 가중치를 두어 유방염(mastitis)에 대한 유전체 예 측을 진행한 결과, GBLUP보다 약 3.9%가 증가된 정확도를 보임을 확인하였다[22].
Table 1에서 기술하였듯이 가장 대중적으로 활용되는 유 전체 예측인 GBLUP은 개체 간의 유전적 관계행렬을 사용 하고, 개체 간의 혈통정보가 추가된 single-step BLUP 모 델이 현재까지 가장 기본적인 유전체 예측법으로 이용되고 있지만, 모델에 대한 가정이 단순하고, QTL 또는 Quantitative Trait Nucleotide (QTN)과 같은 생물학적 기능을 고려하지 않고 개체별 유전자형간의 관계만을 고려한다. 그 렇지만, 형질에 연관된 주요한 양적형질좌위가 존재한다면 Bayes Cpi, Bayes R과 같이 SNP 효과의 다양한 분포 및 가중치를 고려하거나, 혼합 모형에 양적형질좌위, 원인 유전 자군 등의 생물학적 기능 정보를 추가하여 분석하는 방법들 이 더 정확한 결과를 얻을 수 있다는 것은 현재까지 연구 결 과들을 종합해보았을 때 국내에도 적용 가능성이 높으며 시 도해보아야 할 것이라고 판단된다. 이러한 접근법은 하나의 유전자 수준에서 대사회로 수준, 더 나아가 각기 다른 멀티 오믹스 자료를 수집하여 범위를 확장해야 하며, 게놈 수준에 서 후성유전체, 전사체, 메타볼롬, 단백체가 표현형을 설명 할 수 있도록 자료 통합을 할 수 있어야 한다. expression Quantitative Trait Loci (eQTL) 연구가 게놈과 전사체가 통합된 하나의 연구방법이다. eQTL은 유전자의 전사를 조 절하거나, 전사하는 부위에 결합하는 단백질과 같이 cisacting 또는 trans-acting의 역할을 하기 때문이다[23]. 국 내에서는 아직까지 유전체예측을 위해 상용칩 자료의 수집 을 통한 유전체 예측, 소규모 집단의 전사체, 후성유전체 연 구가 진행되고 있는 실정이다. 그렇지만 유전체 예측을 위해 다양한 유전체 정보의 통합, 효율적 사용을 위한 자료의 최 적화 등의 연구 현황을 살펴보면 세계적인 연구 추세에 맞 추어 유전체 연구 방향의 범위를 확장하는 것이 필요할 것 이다. 특히, 유전체정보는 측정하기 어려웠던 사료효율, 메 탄가스 방출 형질부터 불가능한 형질(수소에서의 유생산 또 는 생축에서의 도체형질)까지 형질 예측에 적용 가능하다 는 것은 향후 유전체 정보의 효용성은 증가될 것이기 때문이 다. 최근 들어 이슈가 되고 있는 정밀농업은 인간에서 유전 체정보를 기반으로 맞춤형 의학 실현을 목표로 하듯이 가축 에서도 개체별 유전체 정보가 추가되면 개량, 형질예측 효과 가 극대화될 수 있다 [24]. 이를 위해서, 씨 가축 또는 그에 상응하는 주요 개체 또는 집단에 대한 다양한 유전체정보 및 표현형 정보를 축적할 수 있는 인프라와 응용 연구가 수반되 어야 할 것이다.
Conclusion
사람에서는 유전체 예측을 주로 당뇨, 고혈압, 정신질환 등 개인 맞춤 의학을 고려하여 관련된 유전자, 변이 정보들을 추출하여 질병을 사전에 방지하거나, 건강할 수 있도록 하 는 연구에 중점을 둔다. 하지만, 가축에서는 질병을 포함하 여 개체가 보유한 능력을 조기 예측하여 선발하는 연구에 주 로 활용 된다. 작게는 유전체 선발, 크게는 유전체 예측이라 는 범위 안에서 ‘유전체’라는 공통 자료를 사용한다. 따라 서, 유전체 연구분야에서는 유전체 예측의 정확도 향상을 위 해 기하급수적으로 생산되고 있는 다양한 유전체 정보의 특 성을 파악하고, 분석하여 유전체 예측 모델에 적용하여 형질 (표현형)을 예측하는 것이 중요하다. 과거에 유전체정보의 제한적 사용으로 가축의 경우, 상용칩을 사용하여 유전체 예 측 연구를 수행하였지만 최근 3~5년간 염기서열해독비용 이 기하급수적으로 감소하면서 다양한 유전체 정보를 활용 하여 유전체 예측 연구를 진행하여, 모델의 정확도 향상을 위해 연구들이 수행 중이다. 그 결과, 각 형질에 영향을 주는 유전자의 구조와 기능을 예측 모델에 추가, 보정하여 일반 적으로 활용되는 GBLUP을 고도화한 GFBLUP, systems genomic BLUP[25] 등의 용어들이 등장하며 새로운 예측 모델이 개발되고 있다. 유전체 예측에 일반적으로 사용되던 단일염기서열다형성 정보가 확장되어 멀티 오믹스 정보들 이 점차 추가 적용되어 형질에 영향을 주는 유전자 기능, 생 물학적 대사회로 정보까지 활용하게 된 것이다. 뿐만 아니 라, 사람에서는 유전체 예측을 위하여 기계 학습, 인공지능 등 다차원 통계기법 적용 등 다양한 학문이 융합되어 유전체 예측이 진행되고 있다. 향후 가축의 경제형질에 대한 정확한 형질 예측을 위해 다양한 통계학적 방법과 유전체정보의 결 합과 최적화가 필요할 것이며, 분석 목적에 맞는 유전체정보 의 선택 또한 요구될 것이라 사료된다.