Introduction
Malignant peripheral nerve sheath tumors (MPNSTs) are defined as malignant tumors arising from peripheral nerves or differentiating along the line of the elements of the nerve sheath. MPNSTs that originate from the brain parenchyma are exceptionally rare and are termed malignant intracerebral nerve sheath tumors (MINSTs). To date, there have been 17 documented cases [1-17]. However, none of these cases describe the epithelioid variant of MINST. The rare epithelioid variant is characterized by predominantly epithelioid cytomorphology and a multinodular growth pattern [18]. Epithelioid MPNST (EMPNST) differs from MPNST in that EMPNST shows strong and diffuse S-100 protein positivity, shows no association with neurofibromatosis type 1, and occasionally originates from a schwannoma [18]. In this paper, we describe the first case of an epithelioid MINST (EMINST).
Case report
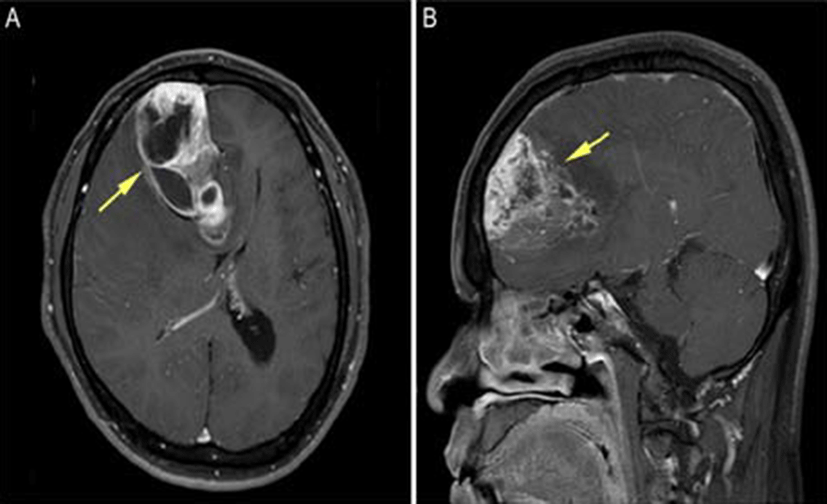
A 50-year-old man was hospitalized as a result of headaches and muscle weakness, which started 6 months previously with new-onset dysarthria. Brain magnetic reso- nance imaging (MRI) revealed a highly enhancing mass approximately 7.7 cm in maximum diameter in the right frontal lobe, crossing the midline via the genu of the corpus callous to the left frontal lobe (Fig. 1A and 1B). Calcification was also identified. Preoperative differential diagnoses included glioblastoma multiforme or a highgrade tumor such as anaplastic oligodendroglioma. A surgical resection was performed through a right frontal craniotomy. Postoperatively, the patient received 6,000 cGy radiation in 30 fractions to the tumor bed. Follow-up brain MRI every 3 months did not show any evidence of tumor recurrence. The patient is currently doing well, 13 months after surgery.
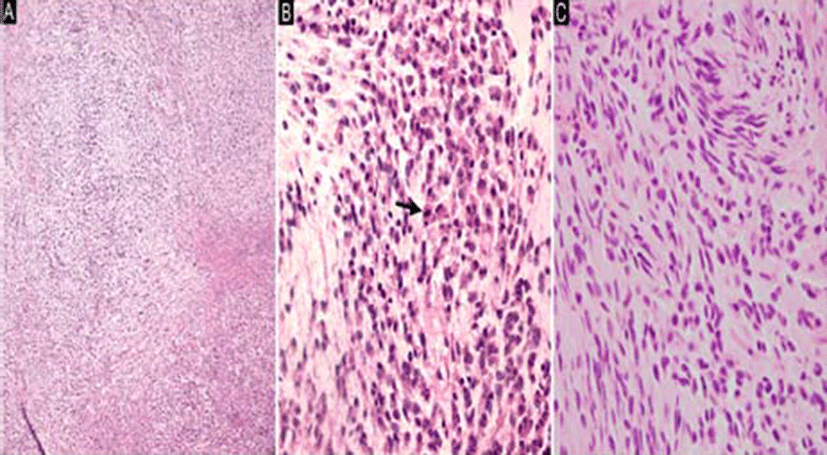
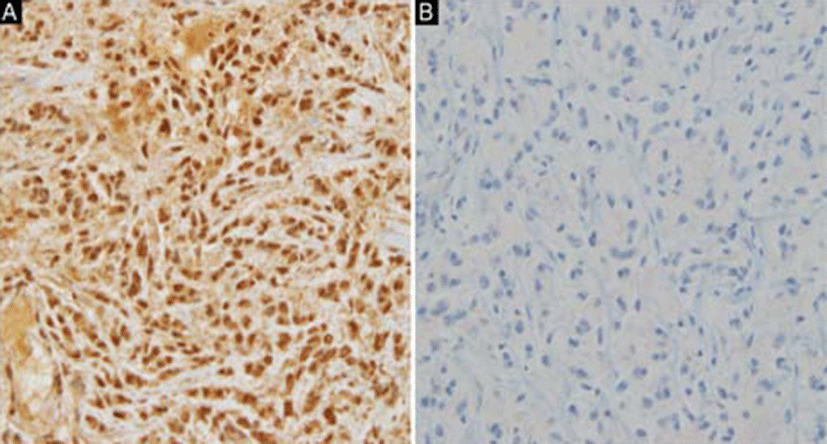
Macroscopically, the tumor was received as multiple fragments of white and yellow soft-to-firm tissue measuring 3.6 × 2.5 × 0.5 cm in aggregate. Microscopically, the tumor cells were arranged in short cords or nests with vaguely nodular patterns embedded in the myxoid stroma (Fig. 2A). Stromal calcification was also noted. Most of the tumor cells had round, polygonal, or ovoid nuclei, moderate amounts of eosinophilic cytoplasm, and indistinct nucleoli (Fig. 2B); however, a few spindle cells were also noted (Fig. 2C). Regarding mitotic activity, 15 mitotic figures were noted per 10 high-power fields (HPFs). Immunohistochemistry (IHC) showed that tumor cells exhibited diffuse immunoreactivity for S-100 (Fig. 3A) and synaptophysin, but were negative for AE1/ AE3, glial fibrillary acidic protein (GFAP), and HMB- 45. Furthermore, INI1 immunostaining was negative (Fig. 3B). The histologic and immunohistochemical findings were consistent with a diagnosis of the epithelioid variant of MINST.
Discussion
MINST is a rare tumor defined as the intracerebral counterpart of MPNST. Both tumors share the same his- tologic and immunohistochemical characteristics of spindle cells with a fascicular growth pattern and focal S-100 immunopositivity [4]. Since the first report by Bruner et al. [17] in 1984, 17 cases of MINST have been reported. However, there are no previous reports of the epithelioid variant of MINST. The epithelioid variant of this tumor is a distinct subtype defined as predominantly epithelioid cells with a multilobular growth pattern and diffuse S-100 positivity.
Clinical features of all previously described MINST cases are summarized in Table 1. The majority of cases were located in the cerebrum, while a few exhibited cerebellar and intraventricular involvement. In the present case, the tumor was intraparenchymal (right lobe) and crossed the midline via the genu of the corpus callous to the left frontal lobe. As shown in Table 1, MINSTs were slightly more common in males (58.8%) than females, and 9 patients (52.9%) were children (age 18 or younger). Most patients were treated by gross total resection; 11 (64.7%) received radiotherapy and/or chemotherapy. Follow-up data were available for 16 cases (range, 0.33 to 77 months; mean duration, 20.7 months). Five (31.3%) patients had no evidence of disease at the time of follow-up. Similar to MPNST, the prognosis of conventional MINST is generally poor with varying risks for local recurrence and metastasis (Table 1) [5, 9, 14, 16, 17]. There are no reports on the prognosis of EMINST, although considering the fact that approximately half of EMPNST patients reported in the literature died of distant metastases, EMINST may also result in a poor outcome [18]. Our patient did not show any evidence of recurrence or distant metastases throughout the followup period (13 months). EMPNST is not associated with neurofibromatosis type 1 and may occasionally originate from a benign schwannoma. Our patient had no history of neurofibromatosis or a pre-existing benign schwannoma.
| Reference | Age (years) /gender | Site | Treatment | Recurrence (months) | Follow-up (months) | Survival (at last F/U) |
|---|---|---|---|---|---|---|
| Lee et al.[1] | 13/M | Right frontal | GTR + CT + RT | 50 and 54 | 77 | alive |
| Shweikehet et al.[2] | 18/M | Right fronto-parietal | GTR+RT | 44 | 52 | dead |
| Ellis et al.[3] | 9/F | Right fronto-temporal | STR + CT + RT | No | 6 | alive |
| Barnard et al.[4] | 75/F | Left frontal | GTR + RT | No | 12 | alive |
| Oztanir et al.[5] | 1/F | Right | STR | NA | 1.5 | dead |
| frontotemproparietal | ||||||
| Kozic et al.[6] | 39/M | Left intrapontine | Biopsy | NR | NR | NR |
| De Cauwer et al.[7] | 68/F | Left parieto-frontal | GTR + RT | 5 | 5 | dead |
| Beauchesne et al.[8] | 35/M | Right cerebral | CT + RT | 17 | 29 | dead |
| peduncle | ||||||
| Maiuri et al.[9] | 36/M | Cerebellar vermis | GTR + RT | 6 | 8 | dead |
| Bornstein-Quevedo et al.[10] | 3/M | Right | STR | NA | 0.33 | dead |
| parieto-occipital | ||||||
| Takahashi et al.[11] | 57/M | Left lateral ventricle | GTR + CT + RT | No | 4 | dead |
| Tanaka et al.[12] | 4/F | Right | GTR | No | 19 | alive |
| parieto-occipital | ||||||
| Sharma et al.[13] | 8/F | Right temporal | GTR | No | 17 | alive |
| Jung et al.[14] | 40/M | Right | GTR + RT | 8 | 8 | dead |
| lateral ventricle | ||||||
| Singh et al.[15] | 61/F | Right cerebellum | GTR + RT | 10 | 18 | dead |
| Stefanko et al.[16] | 15/M | Left parieto-occipital | GTR + CT + RT | 5 and 8 | 9 | dead |
| Bruner et al.[17] | 18/M | Frontal | GTR | 24, 48, | 66 | alive |
| and 66 | ||||||
| Present case | 50/M | Right frontal | GTR | No | 13 | alive |
Table 2 shows the pathologic findings of all previous cases of MINST. Five (29.4%) cases reported the presence of focal epithelioid cells, although of a lesser amount than spindle cells, and therefore do not fulfill the definition of the epithelioid variant. In the present case, most of tumor cells were epithelioid type, which could be difficult to diagnose, however, the presence of a few foci of spindle cells admixed with epithelioid cells raised the possibility of MINST of epithelioid variant. Furthermore, the multilobular growth pattern of our case is different from the fascicular pattern of 17 other cases of conventional MINST. The other features of malignant tumors, including nuclear pleomorphism, mitotic activity, and necrosis, were variable. There is no definite asso- ciation between these pathologic features and outcomes. Our case showed moderate nuclear atypia and a mitotic rate of 15/10 HPFs with necrosis. The IHC results for S-100 protein in the 17 cases of conventional MINST were inconsistent.
| Reference | Tumor size (cm) | Pattern | Composed cells | Nuclear pleomorphism | Mitotic activity | Necrosis | S-100 |
|---|---|---|---|---|---|---|---|
| Lee et al.[1] | 6.7 | Interwoven and | Spindle cells | NR | Moder- | NR | + (focal) |
| interacing fascicles | ate | ||||||
| Shweikehet | 4.1 × 3.5 × 4.1 | Fascicles | Spindle cells | + | Nu- | + | - |
| et al.[2] | merous | ||||||
| Ellis et al.[3] | 8 × 6.5 × 7 | Fascicular | Spindle and | NR | >10/10 | NR | + (faint, |
| epithelial cells | HPFs | diffuse) | |||||
| Barnard et al.[4] | 6.8 × 5.0 × 4.6 | Interlacing | Spindle cells | + | + | NR | + (sparce) |
| short fascicles | |||||||
| Oztanir et al.[5] | 8 × 8 × 8 | Fascicular | Spindle cells | NR | A b u n - | + | + (strong, |
| dant | diffuse) | ||||||
| Kozic et al.[6] | 3.0 × 3.2 | Fascicular | Spindle cells | NR | NR | NR | + |
| De Cauwer et al.[7] | Spindle cells | ||||||
| NR | NR | with rhabdomatoid | NR | High | + | + | |
| differentiation | |||||||
| Beauchesne | 2.2 | Fascicular | Spindle cells | + | Scarce | - | + |
| et al.[8] | |||||||
| Maiuri et al.[9] | NR | NR | Spindle and | + | 4/10 | NR | + |
| epithelioid cells | HPFs | ||||||
| Bornstein- Quevedo et al.[10] | Spindle and | 8/10 HPFs | |||||
| 5.1 3.2 | Fascicular | rhabdomyoblastic | + | + | + | ||
| cells | |||||||
| Takahashi et al.[11] | Spindle and | ||||||
| 3.0 × 4.0 × 3.0 | Herringbone | rhabdomyoblastic | NR | + | - | ||
| cells | |||||||
| Tanaka et al.[12] | Interlacing | NR | |||||
| 5.0 × 5.0 × 4.0 | fascicles, | Spindle cells | NR | 10/10 HPFs | + (intense, focal) | ||
| cystic components | |||||||
| Sharma et al.[13] | 3.4 × 2.7 × 0.4 | Intertwining | Spindle and | + | 3/10 | NR | + (intense, |
| fascicles | epithelioid cells | HPFs | diffuse) | ||||
| Jung et al.[14] | 5.0 × 5.0 × 6.0 | Interlacing | Spindle cells | NR | Fre- | NR | + (diffuse) |
| fascicles | quent | ||||||
| Singh et al.[15] | NR | Interwoven | Spindle cells and | + | Many | + | + (uniform) |
| fascicles | epithelioid cells | ||||||
| Stefanko et al.[16] | 6.2 × 6.2 | Fascicles | Spindle and | + | High | - | + (only epithe- |
| epithelioid cells | lioid cells) | ||||||
| Bruner et al.[17] | NR | Fascicles | Spindle and | + | Rare | - | + (uniform) |
| epitheliod cells | |||||||
| Epithelioid cells | + | ||||||
| Present case | 7.7 | Multilobular growth | and | + | 15/10 HPFs | + (intense, diffuse) | |
| a few spindle cells |
By contrast, our case showed diffuse and strong S-100 positivity. The diagnosis of MINST is complicated because the cellular origin of this tumor has been unrevealed [4, 8, 15]. However, considering the histology and IHC results mentioned so far, the present case could be diagnosed as epithelioid variant of MINST.
Recently, Hornick et al.[19] and Jo et al.[18] reported the loss of the tumor-suppressor gene product SMARCB1/ INI1 in EMPNST, which was recognized in 12 of 24 cases (50%) and 35 of 52 cases (67%), respectively. The present case also shows loss of INI1 expression as determined by IHC. By contrast, conventional MPNST does not show decreased INI1 immunoreactivity [18]. Therefore, EMPNST differs from conventional MPNST both morphologically and genetically. The genetic mechanism of INI1 loss in EMPNST has not yet been elucidated. However, Carter et al.[20] reported a novel germline mutation (c,245_246insAT) in the SMARCB1/INI1 gene in a patient with an EMPNST arising within a “neuro blastoma-like” schwannomatosis. The significance of the functional loss of SMARCB1/INI1 in tumor formation is unclear, although a recent report suggests that loss of SMARCB1/INI1 results in aberrant activation of the hedgehog pathway, contributing to tumorigenesis [20].
EMINST should be distinguished from epithelioid schwannoma. The histologic findings of the latter are characterized by epithelioid cytomorphology with variable amounts of amphophilic to eosinophilic cytoplasm with a multilobular growth pattern and a frequently fibrous and myxoid stroma, which is reminiscent of EMPNST at low-power magnification. Furthermore, both tumors show diffuse immunopositivity for S-100 protein [18]. However, there are no reports of epithelioid schwannoma in an intracerebral location. In fact, epithelioid schwannomas are mostly dermal or subcutaneous in location, with well-circumscribed margins and a lack of significant cytologic atypia [18]. The presence of nuclear pleomorphism, vesicular nuclei, and atypical mitotic figures in our case could differentiate EMINST from epithelioid schwannoma.
Amelanotic melanoma, whether primary or metastatic, is another important differential diagnosis of EMINST. Malignant melanoma can occur at any anatomic site and shows similar histologic findings of epithelioid cytomorphology, a nested growth pattern, and diffuse S-100 immunopositivity, probably due to the similar embryologic origins of melanocytes and Schwann cells in the neural crest [18]. However, the immunoprofile of malignant melanoma is different from that of EMINST in that malignant melanoma shows intact INI expression as well as positive melanoma-associated marker including HMB- 45 [18]. Furthermore, the present tumor was positive for synaptophysin. Moreover, malignant melanoma typically lacks a myxoid stroma, which is frequently seen in EMINST and shows greater cytologic atypia.
Given the intracerebral location and MRI findings, glial tumors such as epithelioid glioblastoma or anaplastic oligodendroglioma could be included in the differential diagnosis. Epithelioid glioblastoma also shows a relatively large, round, and abundant eosinophilic cytoplasm with a lack of cytoplasmic processes and often displays large nuclei with single prominent nucleoli. However, this differential diagnosis was excluded by the absence of GFAP immunopositivity. Anaplastic oligodendroglioma is a diffusely infiltrating glioma composed of cells with rounded nuclei and focal microcalcification. However, a perinuclear halo and branching capillaries were not observed in our case. Also, the epithelioid cytomorphology and cord-like or nested growth pattern were not compatible with oligodendroglioma. In addition, negative GFAP and positive S-100 and synaptophysin immunostainings are more favorable for the diagnosis of EMINST.
Although less likely, a metastatic carcinoma was also considered. However, our patient had no history of primary carcinoma elsewhere, and tumor cells were less cohesive with negative immunostaining results for AE1/ AE3.
In conclusion, we report a case of the epithelioid variant of MINST. There are prior reports of MINST with a focal epithelioid component; however, these are not compatible with the definition of EMINST, which is predominantly composed of epithelioid cells. To our knowledge, our case is the first report of the epithelioid variant of MINST thus far.