Introduction
There are about 17 Mollicutes species that can be isolated from swine. Some of them are recognized as pathogenic, but others may be present in an occasional way with unknown pathogenic capabilities [1]. Mycoplasma hyopneumoniae, Mycoplasma hyorhinis, Mycoplasma hyosynoviae, and Mycoplasma flocculare are highlighted due to their importance and ability to cause infections related to pneumonic processes in the respiratory track, joints, eyes, and genital track [2].
M. hyorhinis, a major contaminant of mammalian tissue cultures in laboratories worldwide, usually infects pigs by causing respiratory tract disease and inflammation of the chest and joints [3]. Polyserositis, arthritis, pneumonia, otitis, and conjunctivitis are clinical disorders associated with M. hyorhinis infection [4]. In porcine respiratory disease complex, M. hyorhinis appears to be frequently associated with M. hyopneumoniae, the primary agent of enzootic pneumonia [5]. Respiratory diseases remain the most challenging health problem in pig production worldwide [6]. For example, in France, a study detected pneumonia in 72.4% of slaughter pigs and pleuritic in 14.4% [7]. These lung diseases result in financial losses due to poor growth performance, reduced feeding efficiency, and higher medication costs and have an adverse effect on pig welfare [6]. Moreover, administration of antimicrobials may have potential negative effects on human health when associated with food-borne contamination by resistant pathogens (even if M. hyorhinis is generally susceptible to antibiotics used against M. hyopneumoniae) or resistant commensal bacteria [8].
Diagnosis of this microorganism is carried out by microbiological culture, serology, and molecular techniques such as polymerase chain reaction (PCR) [9]. M. hyorhinis can be isolated from live pigs (nasal, oropharyngeal, and tracheal swabs or oral fluids) as well as dead pigs (tissue or swab samples taken from synovial fluid of arthritis cases or pleural, pericardial, and peritoneal lesions) [5, 9]. Isolation of M. hyorhinis is relatively tedious and time-consuming. Colonies can be observed on solid media around 4 to 15 days after inoculation [9]. Difficulty in culturing M. hyorhinis has led to the development of other diagnostic assays. It is necessary to clarify the current status of Mycoplasma contamination in animal colonies, as they are prevalent in commercial and animal facilities [10-12]. PCR provides a powerful technique of identifying Mycoplasmas and studying the homology between their nucleic acids [13].
In this study, we aimed to develop a sensitive and specific PCR assay to detect M. hyorhinis and applied the developed PCR assay for detection of Mycoplasma infection in clinical piglets infected with M. hyorhinis.
Materials and Methods
M. hyopneumoniae (ATCC strain 25934), M. hyorhinis (ATCC strain 27717), Mycoplasma pneumonia (ATCC15531), M. pulmonis (ATCC 19612), M. hominis (ATCC23114), and M. arthritiditis (ATCC19611) were obtained from the American Type Culture Collection (Rockville, MD). Mycoplasmas were grown in modified Friis medium [3] containing 20% porcine serum (Gibco-BRL), 5% fresh yeast extract (Gibco-BRL), methicillin (0.15 mg/mL; Sigma-Aldrich Canada, Oakville, Ontario, Canada), bacitracin (0.15 mg/mL; Sigma-Aldrich), and thallium acetate (0.08 mg/mL; Sigma-Aldrich). Cells were harvested by centrifugation at 12,000 g for 30 min at 4°C, washed three times, and suspended in 0.1 M phosphate-buffered saline (PBS, pH 7.4).
We acquired 2-week-old male Landrace piglets from Farmstory Farm (Jeongeup, Korea). The animals were all seronegative against M. hyorhinis. The piglets were acclimatized and kept in a small animal facilitate room with a regulated temperature (23°C ± 1°C), humidity (50% ± 5%), and light/dark cycle (12/12 hr). The animals were fed piglet diet and water ad libitum. After acclimation for 1 week, the animals were divided into two groups (infected and control). Three piglets (piglets No. 1 through No. 3) were experimentally infected with 5 × 109 colony-forming units (CFU) of M. hyorhinis (ATCC strain 27717) (in 1 mL of culture medium) by intranasal inoculation. Three non-infected control piglets (piglets No. 4 through No. 6) housed separately from the infected piglets were used as controls. The animals were monitored clinically for a 4-week period and then euthanized by intravenous administration of pentobarbitone (1.2 g/kg). Their lungs were aseptically collected and processed for the M. hyorhinis-specific PCR assay developed in this study. All studies were performed in accordance with the Guide for Animal Experimentation by Wonkwang University and approved by the Institutional Animal Care and Use Committee of Wonkwang University (Approval No. WKU11-39). All efforts were made to minimize the pain or discomfort of the animals.
DNAs were extracted from cultures of M. hyorhinis (ATCC strain 27717) The lung tissues were homogenized and resuspended in PBS and submitted to DNA extraction as described previously [14]. Briefly, genomic DNA was isolated using an AccuPrep Genomic DNA extraction kit (Bioneer Corporation, Daejeon, Korea) according to the manufacturer's instructions. The DNA was eluted in Tris-EDTA buffer (pH 8.0), and an aliquot was used for PCR amplification. All DNA samples were stored at –20°C until the PCR assays were performed.
M. hyorhinis-specific primer pair, Mrhin-F and Mrhin-R, was designed from the Mycoplasma 16S-23S rRNA internal transcribed spacer (ITS) region sequence for the detection of M. hyorhinis. Forward primer Mrhin-F was 5'- CT TCG GAG ACC ATT GCC TAA G-3' (22 mer, nucleotide 19731~19750). Reverse primer Mrhin-R was 5'- AAA GCT CTC AAA ACT AGA CAC GAA-3' (24 mer, nucleotide 19889~19866).
DNAs were extracted from cultures of M. hyopneumoniae (ATCC strain 25934), M. hyorhinis (ATCC strain 27717), M. pneumonia (ATCC15531), M. pulmonis (ATCC 19612), M. hominis (ATCC23114), and M. arthritiditis (ATCC19611). Amplification of the M. hyorhinis-specific gene was performed with the primer pair Mrhin-F and Mrhin-R, which was designed from the Mycoplasma 16S-23S rRNA ITS gene. The template DNA (50 ng) and 20 pmoL of each primer were added to a PCR mixture tube (AccuPower PCR PreMix; Bioneer Corp., Korea) containing 2.5 U of Taq DNA polymerase, 250 μM of each deoxynucleoside triphosphate, 10 mM Tris-HCl (pH 8.3), 40 mM KCl, 1.5 mM MgCl2, and the gel loading dye. The volume was adjusted with distilled water to 20 μL. The reaction mixture was subjected to denaturation at 94°C for 5 min, followed by 30 cycles of 95°C for 5 min, 62°C for 1 min, and 72°C for 1 min and a final extension step of 72°C for 3 min, and samples were kept at 4°C until analysis. Reactions were conducted using My Genie 32 Thermal Block PCR (Bioneer, Korea). After amplification, a 5 μL aliquot of each PCR was separated by electrophoresis on 2% agarose gels, followed by ethidium bromide staining and UV transillumination.
DNAs were extracted from cultures of M. hyorhinis (ATCC strain 27717). To determine the sensitivity of the developed PCR assay, serial dilutions of purified chromosomal DNA from M. hyorhinis strain were tested. Amplification of the M. hyorhinis-specific gene was performed using the primer pairs designed from the Mycoplasma 16S-23S rRNA ITS gene. After amplification, a 5 μL aliquot of each PCR was separated by electrophoresis on 2% agarose gels, followed by ethidium bromide staining and UV transillumination.
To evaluate the PCR system under field conditions, pulmonary tissues with pneumonic lesions were collected from the piglets experimentally infected with M. hyorhinis. The pulmonary tissues were homogenized, resuspended in PBS, and subjected to DNA extraction, as described previously [14]. M. hyorhinis-specific PCR was performed using the primer pairs designed from the Mycoplasma 16S-23S rRNA ITS gene. After amplification, a 5 μL aliquot of each PCR was separated by electrophoresis on 2% agarose gels, followed by ethidium bromide staining and UV transillumination.
Results
PCR assay using M. hyorhinis-specific primer pairs was newly developed and evaluated for its specificity. For the PCR amplification, 140 bp amplicons were detected in the extracted DNAs from M. hyorhinis (Fig. 1). The targeted 140 bp region of the Mycoplasma 16S-23S rRNA ITS gene was specifically amplified by the optimized PCR system using the M. hyorhinis-specific primer pairs designed in this study. The specificity of the developed M. hyorhinis-specific primer pair was confirmed using other bacterial DNAs with high homology in their sequences (Fig. 1). No positive signals were observed in the template DNA samples of M. hyopneumoniae (ATCC strain 25934), M. pneumonia (ATCC15531), M. pulmonis (ATCC 19612), M. hominis (ATCC23114), and M. arthritiditis (ATCC19611) (Fig. 1). However, DNAs of M. hyorhinis resulted in a strong positive signal (Fig. 1).

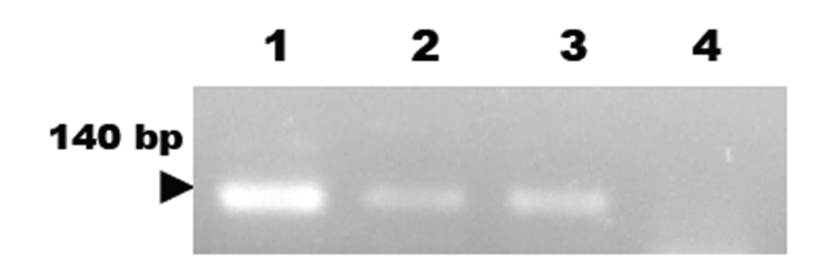
To evaluate the sensitivity of the developed M. hyorhinis-specific PCR assay, serial dilutions of DNA extracted from cultured M. hyorhinis were tested with our PCR assay. As a result, we detected at least of 1 pg of genomic DNA in 5 μL aliquots of PCR product by ethidium bromide staining (Fig. 2).
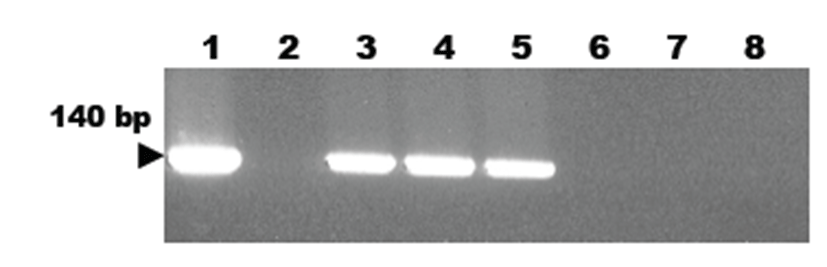
A new developed PCR method using primers designed from the Mycoplasma 16S-23S rRNA ITS gene sequence was employed to detect M. hyorhinis infection in piglets. As a result, the targeted 140 bp gene was specifically amplified by the developed PCR method using the M. hyorhinis-specific primer pair. The target nucleic acid fragments were specifically amplified in lung tissue samples of M. hyorhinis-infected pigs using the developed PCR method (Fig. 3). However, we did not detect any positive signals in tissue samples of non-infected normal pigs (Fig. 3).
Discussion
M. hyorhinis is considered an etiological agent of arthritis in sucking pigs [3]. However, inoculation studies on this microorganism in young gnotobiotic pigs showed that some M. hyorhinis strains produce mild catarrhal pneumonia with gross and microscopic lesions that are indistinguishable from the mycoplasmosis caused by M. hyopneumoniae [15]. As already mentioned, M. hypneumoniae has been historically recognized as the major etiological agent in pig pneumonias [15, 16]. However, M. hyorhinis strains with a set degree of virulence were sufficient to cause lesions in lungs [17]. These findings suggest that in chronic pneumonia, concomitant infection by M. hyorhinis may increase lesions of purulent bronchopneumonia responsible for causing pleurisy [17].
M. hyorhinis is also an infectious agent potentially implicated in human cancers. Accumulating evidence suggests that M. hyorhinis infection in humans results in clinical outcomes [18-20]. Working independently, several groups have detected M. hyorhinis p37 protein in cancer patients [21-24]. p37 is a peptide of 403 amino acids with a molecular mass of 43.5 kDa. Analysis of the protein sequence revealed that p37 has about 34% to 42% similarity to the periplasmic binding protein-dependent transport systems found in Gram-negative bacteria and several species of Mycoplasma. Thus, p37 is thought to be part of a high-affinity transport system in M. hyorhinis [21].
Diagnosis of Mycoplasma species is usually done by cultivation of the organism or by immunofluorescence tests performed on frozen thin lung sections with polyclonal antibodies [4,11]. However, due to the fastidious nature of Mycoplasma species, its culture and serological identification may take up to 1 month. Due to advances made in molecular biology over the last few years, more is known about Mycoplasma species genes. Hence, other methods can be used as diagnostic tools for this organism. Recently, PCR methods have been used to detect Mycoplasma species [25]. In order to circumvent those limitations, many nucleic acid technology-predicated procedures have been developed. PCR-based methods for the detection of certain DNA regions in the Mycoplasma genome have proven to be both rapid and specific [26-30].
Sequencing methods have been introduced as a new way to study the molecular epidemiology of bacterial pathogens [1]. Among these methods, sequencing of the 16S-23S rRNA ITS region is a new weapon for epidemiological studies. As this region can be variable in sequence and size in different species of Mycoplasmas, it has been used to compare microorganisms of the same species or among species with a close phylogenetic relationship [31].
In our case, amplification of the 16S-23S rRNA ITS region of M. hyorhinis was performed, and PCR results are shown in Figure 1, where variability between strains of the same species is observed. In this study, we developed a new PCR assay using a M. hyorhinis-specific primer pair, Mrhin-F and Mrhin-R. The primers and probe for the assay were designed from the 16S-23S rRNA ITS region of the Mycoplasma rRNA unique to M. hyorhinis. The developed PCR assay was very specific and sensitive for the detection of M. hyorhinis. The assay could detect the equivalent of 1 pg of target template DNA, which indicates that the assay was very sensitive. In addition, the M. hyorhinis-specific PCR assay detected only M. hyorhinis and not any other Mycoplasma spp. Further, the new developed PCR analysis effectively detected M. hyorhinis infection in pigs.
We suggest that this PCR assay using the M. hyorhinis-specific primer pair Mrhin-F and Mrhin-R could be useful and effective for monitoring M. hyorhinis infection in pigs.