Introduction
The canine distemper virus (CDV) belonging to Paramyxoviridae affects the families of dogs and weasels which produce canine distemper disease [1]. The virus enters through the respiratory organ and reproduces in the lymphatic tissues of the respiratory system. After four to five days of incubation period, the infected dogs experience heat, tears, runny nose, vomiting, viscous diarKyong rhea, and nervous symptoms such as stiffening and convulsions of the soles and nose [2]. The lethality of young dogs is usually high because it involves complications such as pneumonia and encephalitis [3]. The viruses are cultured and proliferated in the chicken embryos and various cell lines. The Madin-Darby canine kidney (MDCK) cells that have been established from adult female dogs are known to have susceptibility to CDV [4].
Canine distemper vaccine was firstly developed by Puntoni. The commercial vaccine was first used in a limited capacity in 1950 [5]. CDV is commonly obtained through cultivation of lymphocytes from infected dogs. The Onderstepoort vaccine strain to which the cell culture has been introduced can be proliferated in various cell strains [6]. The Rockbone vaccine strain was separated by cultivating in canine kidney cells. More recently, recombinant vaccines were developed which use hemagglutinin protein and modified RNA polymerase as immunogen [7].
To culture animal cells, the tissue slices removed from specific organs or tissues are grown in aseptic condition. The cells obtained from this process are called primary culture cells. The primary cells changes its characteristics after passing through freezing and thawing and have the disadvantage of difficulty in using for a long time because of the small valid passage count [8]. However, unlike cells produced from cancer tissues or cells that have passed through canceration, primary culture cells retain the original characteristics of general cells and can be used in a variety of studies. At present, only a few primary culture methods obtaining cells from embryos are available. Experiments using the embryos of animals have the advantage of using cells for a long time because cell division is active [9, 10]. Furthermore, as there are epithelial cells and fibroblasts in anchorage-dependent cells, separating and using only the epithelial cells during the primary culture is important [11]. The reason is that, when fibroblasts are included, the epithelial cells are disappeared early because the division of fibroblasts is more dominant [12]. In this case, if collagenase and dispase enzymes are appropriately treated, separating only the fibroblasts and obtaining the epithelial cells are possible.
To prevent the inclusion of fibroblasts, edetate disodium dehydrate (Na2EDDA) was used, which disconnects the link of calcium ions (Ca2+) used in the bonding of fibroblasts. As epithelial cells have fewer bonds among Ca2+ compared with fibroblasts, they can be effectively removed by disconnecting only the links of fibroblasts through appropriate treatment. Fetal bovine serum (FBS), which is used in cell culture, gives the attachment factor used for the attachment of cells to the culture flask [13]. As fibroblasts need a greater amount of attachment factors, if this amount is small, fibroblasts are removed because they cannot be attached. Therefore, FBS can be used to obtain only the epithelial cells [14, 15, 16].
This experiment was conducted to develop host cell lines for the high titer production of CDV. Immortalized cell lines were prepared by over-manifesting telomerase (human telomerase reverse transcriptase, h-TERT) in primary canine cells using retrovirus vector. The susceptibility of these cell lines to the CDV was demonstrated.
Materials and Methods
For organ extraction, canine embryos were obtained from a pregnant dog (mongrel) through hysterectomy. The embryos were extracted through an aseptic process from the womb. First, the surface of the womb was sterilized with 70% ethanol. Then, the womb was incised, the umbilical cord was tied by suture, and only the embryo was extracted. The surface of the extracted embryo was sterilized with 100 mg/mL of povidone iodide solution and cleaned with chlorhexidine solution before the kidney was extracted. The MDCK cell line (KCLB no. 10034) for this experiment was bought from the Korean Cell Line Bank (Seoul, Korea). For the virus, CDV (3CL DEF4) from CAVAC (Dajeon, Korea) was used. All animal experiments were approved under the protocol of the Jeju National University Institutional Animal Care and Use Committee (Accreditation No. 2014-0010) and performed in accordance with the guidelines for the Care and Use of Laboratory Animals of the Jeju National University to minimize animal suffering. Animal suffering was reduced by keeping the number of animals used to the minimum required for the planned studies to give statistically significant data.
After the Dulbecco’s modified essential medium (DMEM: GIBCO, New York, USA) was injected, the extracted organs were cut into pieces as small as possible in a glass Petri dish. To obtain the tissue only and remove the RBCs, the pieces were moved to a 50 mL conical tube and allowed to sink for 2 min before the supernatant was removed. Exactly 30 mL of DMEM medium was added to the conical tube, and the sunken small pieces of organ were floated through pipetting. The pieces were left for about 30 sec before the supernatant was removed again. This process was repeated three times to remove the RBCs.
To separate into single cells, the organ was enzyme treated. After the RBCs were removed, the small pieces of the canine kidney were treated with 0.25% collagenase for 20 min at 37°C. Next, a second treatment was performed using Trypsin/EDTA (0.01%, 2 min). Pipetting was performed slowly about 20 times until the pieces were separated into single cells. Then, 30 mL of DMEM with 10% FBS (GIBCO) was added to the cells, which were then centrifuged for 3 min at 160 × g. Single cells were injected in units of 1 × 104 cells to a 24-well cell culture cluster plate. About 1 mL of DMEM with 10% FBS was added to the cell culture cluster plate, and the single cells were cultured for 12 hr in a CO2 incubator at 37°C.
The kidney cells, which were the fourth generation of the primary culture, were cell cultured in a T25 flask for 24 hr until a 70% monolayer was achieved. Phosphate buffered saline (PBS) was used for the cultured cells, which were washed three times. After removing the PBS, 1 mL of 0.05% Na2EDDA was added, and the separation of fibroblasts was observed under a microscope for 2 to 3 min. When the fibroblasts were condensed and fell to the bottom, the flask was lightly patted against one palm to separate the fibroblasts only. The primary culture cells after the removal of the fibroblasts were cultured for 24 hr in a DMEM medium with 2% FBS. Exactly 24 hr after the primary processing, the fibroblasts were removed by the second Na2EDDA processing. Once the fibroblasts were fully removed, DMEM with 10% FBS was added, and the cells were cultured in a CO2 incubator at 37°C.
After h-TRET was amplified in HeLa cells, they were injected into a pDONAI-2 vector and the recombinant retrovirus particles were produced by packing the retrovirus. This process is detailed in [17]. RNA was obtained from HeLa cells using real-time polymerase chain reaction (RT-PCR; Thermoscript reverse transcriptase, Invitrogen, California, USA) and KOD polymerase (TOYOBO ,Osaka, Japan). The following primer was used: 5’ GGA ATT CGC CGC GCG CTC CCC GCT GCC GAG CC 3’ and 5’ GCT CTA GAT TAG TCC AGG ATG GTC TTG AAG TCT 3’. Two clones were combined using Primer 5’ AAA AAG CAG GCT CCA CCA TGC CGC GCG CTC CCC GCT GCC GAG CC 3’ and 5’ AGA AAG CTG GGT TAG TCC AGG ATG GTC TTG AAG TCT 3’. For Adaptor primer, 5’ GGG GAC AAG TTT GTA CAA AAA AGC AGG CT 3’ and 5’ GGG GAC CAC TTT GTA CAA GAA AGC TGG GT 3’ was used. This h-TRET was injected into the pDONAI-2 vector to produce the pDONAI-2-h-TERT vector. Recombinant retrovirus particles were produced from the pDONAI-2-h-TERT vector using the Retrovirus Packing System (TAKARA Bio, Otsu, Japan). The recombinant retrovirus vector (pDONAI-2-h-TERT), pGP vector, and pE-ampho vector were co-transfected in the 293T cell using the calcium phosphate transfection method. After co-transfection, the cells were treated in 5% CO2 at 37°C for 7 to 11 hr before the medium was replaced with a new one. After 48 hr of transfection, the supernatant was removed by centrifuge. After filtering with a 0.45 μm filter, it was kept at –80 °C and used for transduction.
The cells from which fibroblasts were removed after the initial culture and the virus particles were inoculated with multiplicity of infection (MOI) 10 with recombinant retrovirus particles. To sort out the cells to which the gene was successfully introduced, the cells were cultured for about two weeks while adding 500 μg/mL of G418 (geneticin) and replacing the medium every day. The surviving cells were cultured with the amount of G418 reduced in half. The expression and immortalization of telomerase were verified. After being cultured for four weeks, the cells were divided into two types of cell lines: JNUCK-1 for cell lines with large cytoplasm and JNUCK-2 for cell lines with small cytoplasm.
Cells were subcultured and the possibility of 30 or more subcultures was examined. Radioactivity was measured using a scintillation counter for cells that acquired thymidine marked with tritium for each generation. Two T25 flasks were cultured for 4 hr, the culture solution was replaced, and 0.01 mL of the growth factor (Epidermal growth factor: GIBCO, USA) that stimulates cell proliferation was added. After culturing for 1 hr, the tritium label thymidine (0.5 μCi/mL) was added and the medium was cultured for 4 hr to 24 hr. To stop the reaction, 0.01 mL of 1% NaN3 was injected into 0°C ice and PBS was added for washing twice. Then, 0.5 mL of 0.2 N NaOH was added to melt the cells. After about 5 min, the melted cell solution was moved to a test tube, which was washed with 0.5 mL 0.2 N NaOH solution. The cells remaining in the well were recovered. They were placed inside a test tube and neutralized by adding 0.2 mL of 2 N hydrochloric acid. Then, 0.15 mL 5% trichloroacetic acid (TCA) was added, and it was left for 30 min at 4°C for precipitation. Afterwards, the supernatant was removed. The precipitates were floated by 5% TCA and centrifuged for 3 min at 300 × g. The precipitates were moved to a glass filter and washed twice in 5% TCA using ethyl alcohol and acetone. A glass filter was injected into a vial and 10 mL of scintillation cocktail was added. Reactivity was measured with a beta counter.
After their immortalization, the cells were divided into five generations, and the telomerase expression patterns were examined through RT-PCR (iQ5 Real-Time PCR Detection System, Bio-rad, California, USA) using TRAPeze®RT Telomerase Detection Kit and FAM detection (Chemicon international, Massachusetts, USA). RT-PCR was performed by producing TSR8 quantification control, 1,000 cell equivalents of telomerase positive extract control, minus telomerase control, no template control, and experimental sample control by preparing cells in units of 1 × 106 cells for five generations. Three RT-PCRs were performed for each material, and the averages were calculated. The design for RT-PCR is shown in Table 3. The copy number was measured with the following formula and the Plasmid DNA was quantified: Copi/μL = [6.02 × (copy/moL) × amount] [length (bp) × (g/moL/bp).
For the immortalized cell line, the existence of only epithelial cells after the removal of fibroblasts was verified. Moreover, the expression of cytokeratin and fibronectin was verified. In the case of cytokeratin, the epithelial cells can be verified by staining the intermediate filaments that have keratin in the cytoplasm of the epithelial cells. After immortalization, 15 and 30 generation cells were subcultured in a 12-well plate for 48 hr. The cell medium was removed and the cells were washed with PBS. After washing, the cells were treated with 4% paraformaldehyde for 10 min and washed three times with PBS to which 1% BSA was added. After treating for 10 min with PBS to which 0.25% Triton X-100 was added, the cells were washed with PBS three times. After blocking with 0.2% BSA for 30 min, the anti-cytokeratin 18 antibody (Abcam®, Massachusetts, USA) which is a primary antibody diluted at 1:100, was reacted for 2 hr at normal temperature, and the anti-fibronectin antibody (Abcam®), which is a primary antibody diluted at 1:200, was reacted for 1 hr at normal temperature. After the reaction, the cells were washed with PBS three times. The anti IgG FITC (Abcam®), which is a secondary antibody diluted at 1:100, was treated at normal temperature for 45 min. The cells were washed three times. Next, the DAPI that was diluted at 1:3000 was treated for 10 min. The nucleus was stained and washed with PBS and mounted with Prolong® gold antifade reagent (Sigma, California, USA). Then, it was observed with a fluorescence microscope.
MDCK, JNUCK-1, and JNUCK-2 cell lines were cultured in a 96-well plate. The medium was removed when an 80% monolayer was formed, and the monolayer was washed with PBS. Exactly, 50 μL of the seed virus was diluted, and 200 μL of the seed virus was inoculated in each well. The seed virus was inoculated in 1/10 units after diluting in each stage. After incubating in a CO2 incubator at 36°C, the occurrence of cytopathic effect (CPE) was checked. The existence of CPE was checked in each well, and the TCID50/mL was calculated to measure susceptibility.
Results
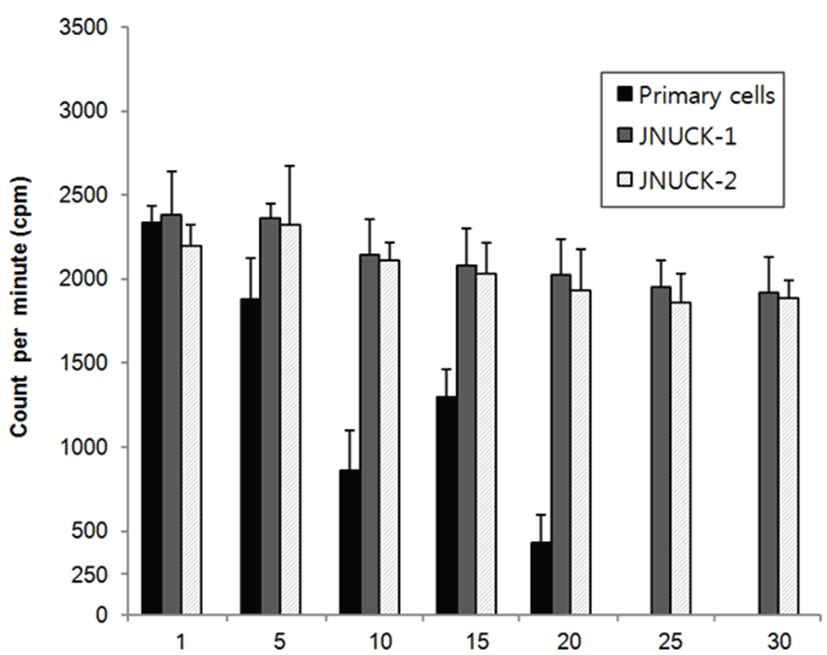
To examine the cell proliferation capacity, tritium-labeled thymidine was injected into the cells that could be cultured until 1, 5, 10, 15, 20, 25, and 30 generations. Their reactivity was measured with a scintillation counter. Among the cells that maintain proliferation capacity through immortalization, the cell line that showed the greatest division multiplication was JNUCK-1 (Fig. 1). The division ability of primary culture cells slowly decreased until the 10th generation and increased in the 15th generation. However, the division ability sharply decreased in the 20th generation. In the 25th and later generations, most cells were killed. The JNUCK-1 and JNUCK-2 cell lines showed division ability of a certain level from the first until the 30th generation.
The maintenance of epithelial cell shape during cell culture, the existence of contact inhibition phenomenon, and multi-layer growth were verified. Furthermore, the disappearance of the characteristics of the epithelial cells after canceration was verified. The produced cell lines were found to grow in a single layer in the form of epithelial cells (Fig. 2).
To sort out the immortalized cells, they were cultured for about 2 weeks while being added with 500 μg/mL of G418 (geneticin) and replaced with the new medium every day. The cells that survived were cultured while being added with G418 at a half concentration, and their immortalization was verified. The immortalized cells were divided into JNUCK-1 and JNUCK-2 by the measurement of the size of the cytoplasm (Fig. 3).
According to the results of the verification of the expression pattern of telomerase, the primary culture cells had the lowest amount of telomerase, whereas both JNUCK-1 and JNUCK-2 cell lines clearly expressed telomerase until the 25th generation (Fig. 4). After three repeated experiments, the cycle threshold values were nearly identical, confirming reproducibility.
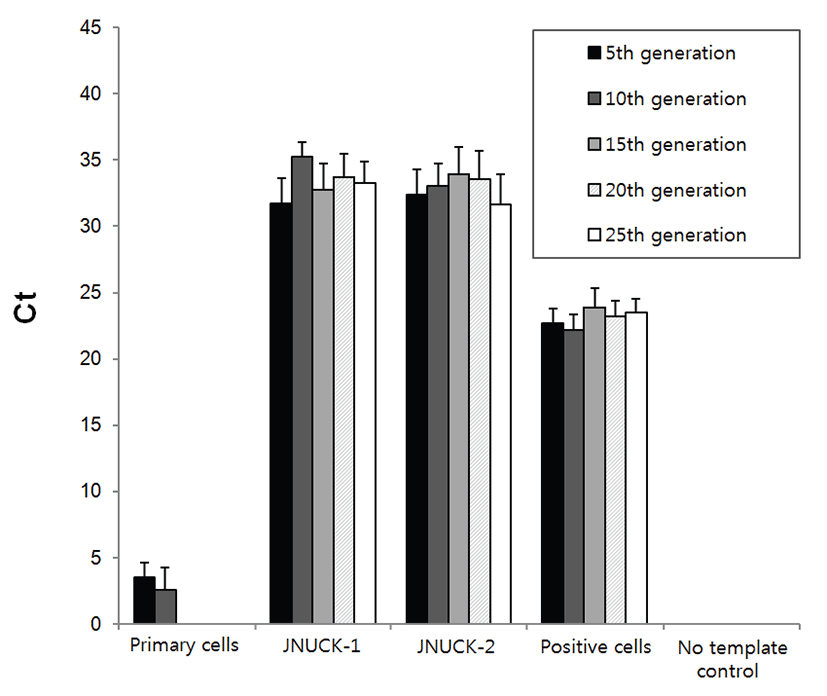
To verify that the immortalized cell lines consisting of epithelial cells, the expression patterns of cytokeratin and fibronectin were verified (Fig. 5). When the primary culture cells were stained with fibronectin, about 60% of the cells were stained. About 20% of the cells were stained when cytokeratin was used for staining. When the JNUCK-1 and JNUCK-2 cell lines were stained with cytokeratin after the removal of fibroblasts, over 90% of the cells secreted cytokeratin. This finding suggests that the fibroblasts were well removed, leaving only the epithelial cells.
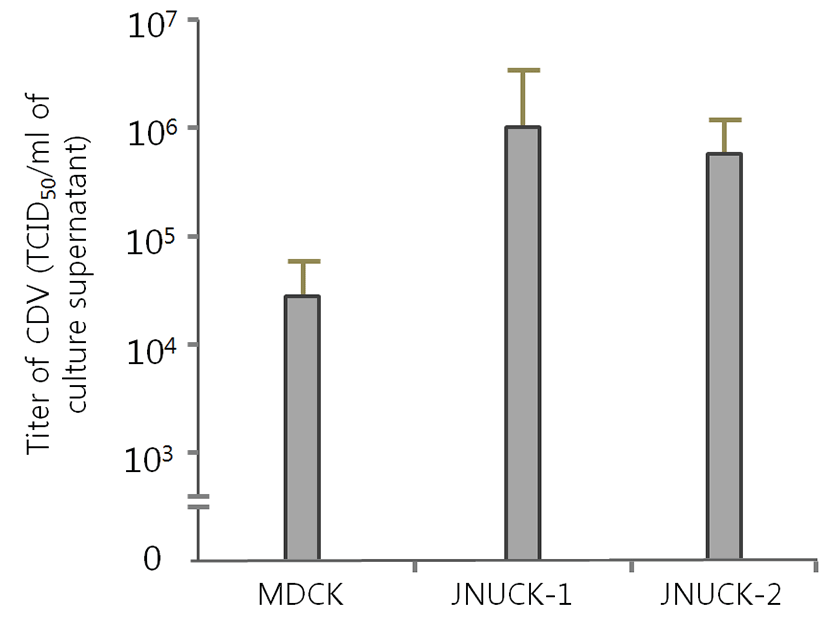
MDCK, JNUCK-1, and JNUCK-2 cell lines were infected with CDV and cultured for seven days. The occurrence of CPE was then verified (Fig. 6). The existence of CPE was verified in each well, and the TCID50/mL was calculated to measure susceptibility. JNUCK-1 showed the highest susceptibility to CVD and JNUCK-2 the lowest (Fig. 7).
Discussion
The CDV used in this study is the type used as the current vaccine strain. At present, the embryo chicken egg is used for the production of CDV for vaccine, but this method has high subculture costs and takes much time [18]. Between the Vero and MDCK cell lines, which are known to be susceptible to CDV, the MDCK cell line is used for virus separation as it is susceptible to CD but not for host cells for mass culture [19]. In particular, the MDCK cell line is derived from canine kidney and is regarded to have high susceptibility and yield compared with other cell lines, but its susceptibility test results are actually low [20]. Therefore, to culture CDV using cell lines, new cell lines need to be produced.
To produce new cells, the kidney cells of canine embryos are primarily cultured. The removal of fibroblasts from the primary culture cells using Na2EDDA requires a certain level of experience and technical know-how [16]. During primary culture, the resistance of fibroblasts to Na2EDDA acts as an important factor as the subculture count increases. To efficiently remove fibroblasts, the Na2EDDA treatment had to be performed before 8 subcultures (about 15 generations) at least [16]. In conclusion, fibroblasts were efficiently removed through primary and secondary Na2EDDA treatments in this study. This experimental method could solve the domination of fibroblasts, which is one of the largest problems in the conventional primary culture, and could be used in various animals and organs.
Various methods for cell production are available, such as direct culture of the cancer cells, production of hybridoma, and knockdown of the P53 gene. However, all these methods have the disadvantage of canceration of all cells. One method for immortalizing cells without canceration is to overexpress telomerase. In this study, the primary culture cells derived from canine kidney were successfully immortalized using the pDONAI-2-h-TERT vector, which is a retrovirus vector (Fig. 4). An examination of the telomerase expression of the produced cell lines through RT-PCR showed a high fluorescence value in both the JNUCK-1 and JNUCK-2 cell lines. Telomerase was expressed continuously until the 25th generation, and the measurement of valid subcultures revealed that the division speed was relatively constant until the 25th generation.
The JNUCK-1 and JNUCK-2 cell lines, the primary culture cells that were not immortalized, which were highly likely to be susceptible to CDV, and the MDCK cell lines that were known to be generally susceptible to CDV were infected with CDV. The CDV susceptibility test results revealed that, after the virus inoculation, the JNUCK-1 cell line showed a higher TCID50/mL than the MDCK cell line (Fig. 7). This finding shows that the immortalization of the kidney JNUCK-1 cell line that was produced in this study maintained the biological characteristics of general cells. Furthermore, the produced cell line showed a titer 200 times higher than that of the conventional MDCK cell line, and it is expected to further develop the early separation and susceptibility test of CDV. Using the produced cell line will be helpful for the fast detection and research of viruses during the early onset of CDV. One of the most important factors in the development of CDV vaccines is the acquisition of cell lines that can be produced at high titers. The cell line developed in this study is appropriate in this regard and appropriate for the production of attenuated virus strains through the continuous subculture of virus. It can also be used as the host cell required for vaccine development. Furthermore, as the cell line produced in this study retains the characteristics of normal epithelial cells rather than cancer cells, it can be used as a host cell line for virus detection or for virus production after a susceptibility test of various dog-derived viruses.